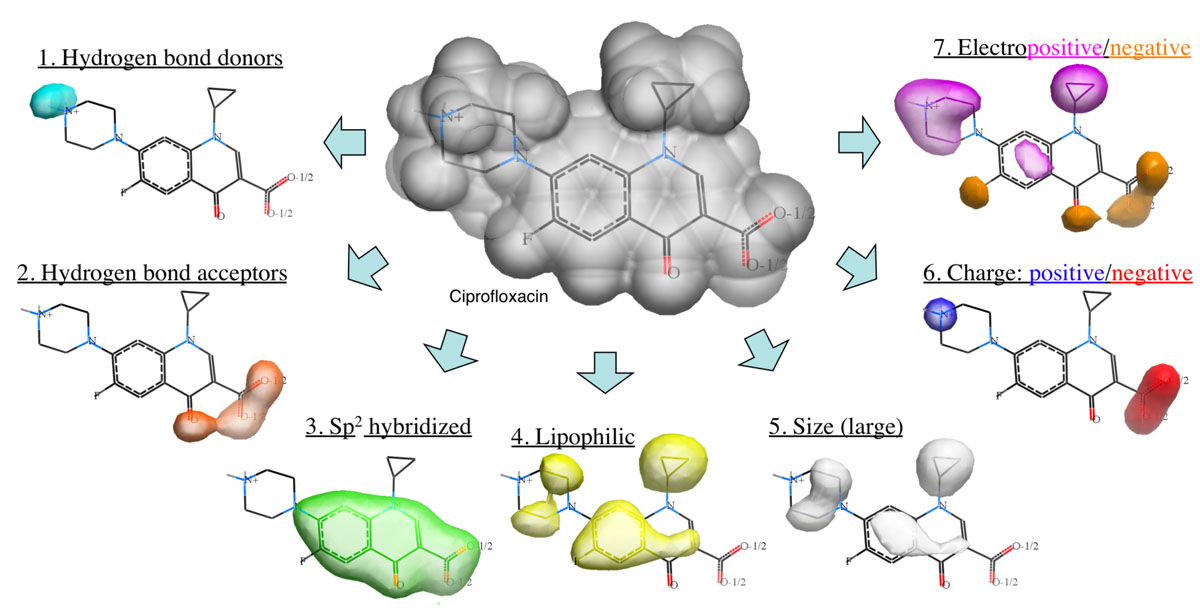
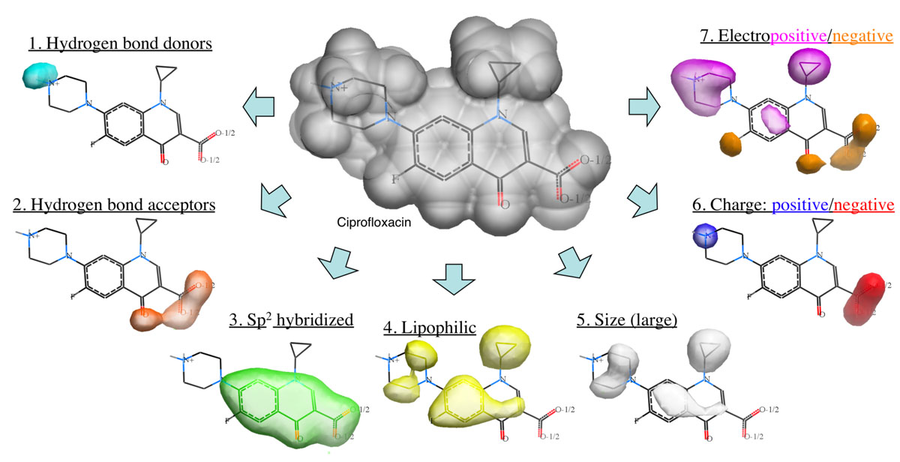
Atomic Property Fields is a grid 3D pharmaophore potential which can be used for chemical superposition, screening and scaffold hopping.
Method
The Atomic Property Field (APF) method developed by MolSoft ( Totrov 2008) is a 3D pharmacophoric potential implemented on a continuously distributed grid which can be used for ligand docking and scoring. APF can be generated from one or more high affinity scaffolds and seven properties are assigned from empiric physico-chemical components. These properties include: hydrogen bond donors, acceptors, Sp2 hybridization, lipophilicity, size, electropositive/negative and charge (image above). A single ligand atom can contribute to multiple fields; multiple similar ligand atoms in a spatially consistent location result in a strong pharmacophore signal for their features in this location. APF has also been extended to multiple flexible ligand alignments using an iterative procedure. APF uses Monte Carlo minimization in the atomic property fields potentials in conjunction with standard force-field energies.
Application
APF pharmacophoric potential is implemented as a grid energy term that can be used for the following purposes:
- Chemical superposition
- Multiple chemical alignment
- Virtual screening
- Scaffold Hopping
- APF-ARM (activity regression model)
- Chemical clustering
- Ligand binding site superposition and comparison (Totrov 2011)
Independent Validation
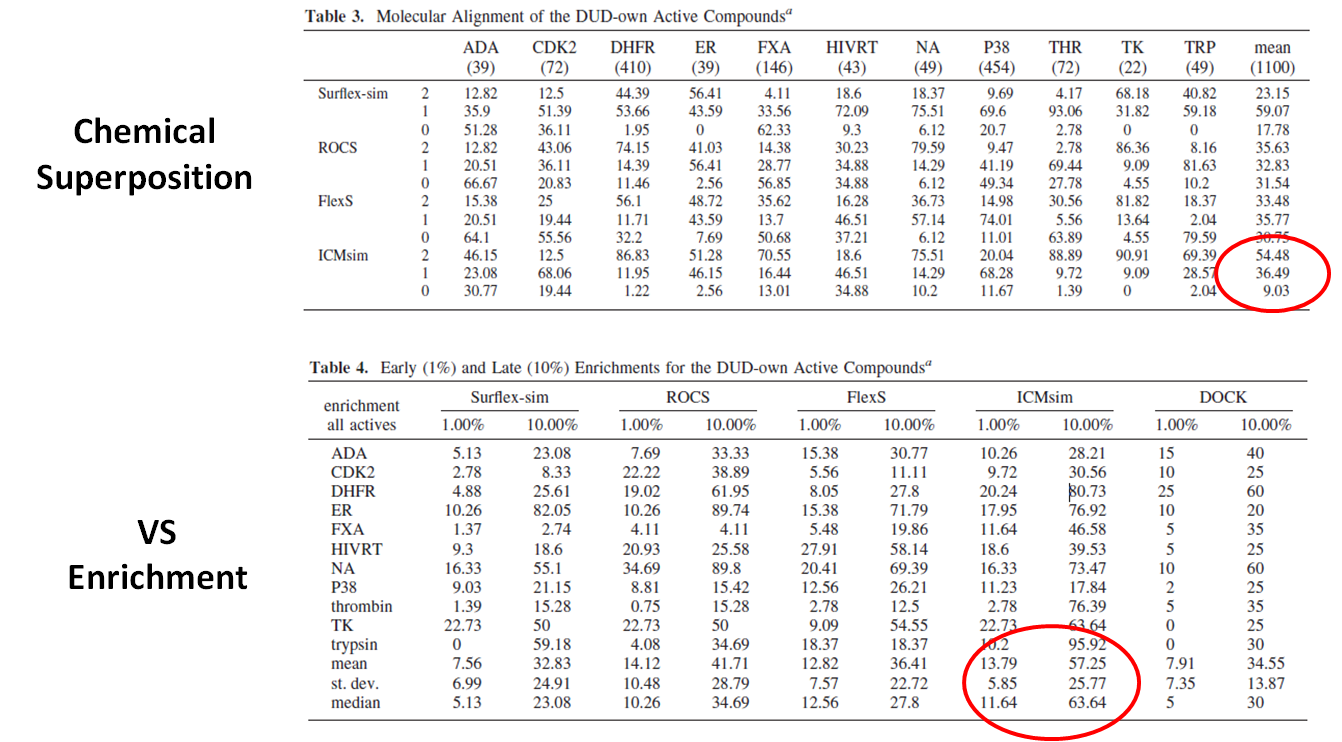
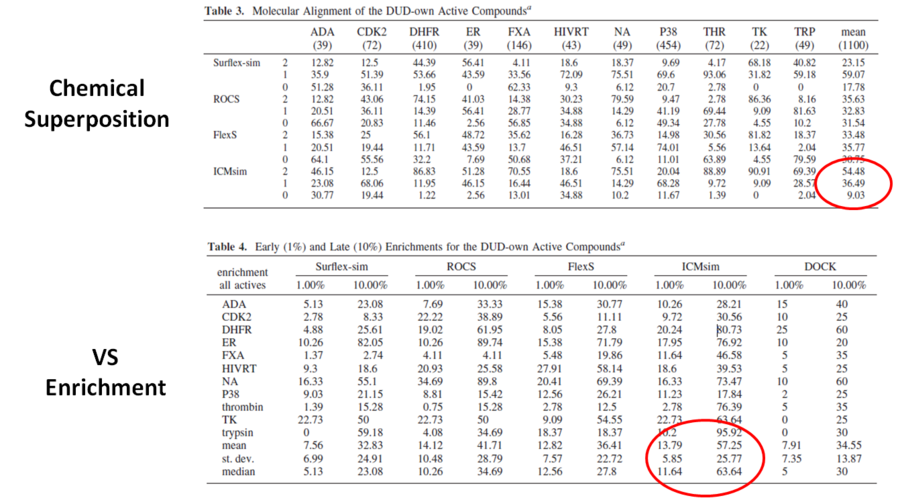
In an independent study, the APF method (Totrov 2008) was shown to be the most accurate method for chemical superposition and ligand-based 3D virtual screening compared to several other commercial and academic software programs (Giganti et al 2010 and see summary in table below). For chemical superposition of active compounds APF (referred to as ICMsim in the paper by Giganti et al) was found to be the most efficient software with an overall acceptable pose rate of 90.97%. For Virtual Screening Enrichment enrichment at 1% APF (ICMsim)performs very well and at later enrichment (10%) APF is significantly better compared to the other programs.
The application of APF screening to nuclear receptors and GPCRs is discussed in a recent publication (Kufareva et al 2013). The APF screening resulted in an excellent recognition of known actives versus property matched decoys with an Area Under Curve (AUC) >0.9 in 19 out of 25 nuclear receptors. This exceeds results obtained using traditional structure-based screening. The majority of the models show a perfect ability to scaffold hop by separating actives from decoys. The test set was designed with sufficient chemical diversity from the training compounds. For GPCRs the results were equally encouraging even when considering the lack of a big 3D training set.
References
Totrov, M. Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem. Biol. Drug Des. 71, 15.27 (2008).
Totrov, M. Ligand binding site superposition and comparison based on Atomic Property Fields: identification of distant homologues, convergent evolution and PDB-wide clustering of binding sites. BMC Bioinformatics 12 Suppl 1, S35 (2011).
Giganti, D. et al. Comparative Evaluation of 3D Virtual Ligand Screening Methods: Impact of the Molecular Alignment on Enrichment. J. Chem. Inf. Model. 50, 992.1004 (2010).
Kufareva, I., Chen, Y.-C., Ilatovskiy, A. V. & Abagyan, R. Compound activity prediction using models of binding pockets or ligand properties in 3D. Curr. Top. Med. Chem. (2012