 and
and
Web Release Date: December 14,
On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors
and
GDECS Computational Chemistry, AstraZeneca R&D, Mölndal, Sweden, Cancer Discovery, AstraZeneca R&D, Boston, Massachusetts, and Medicinal Chemistry, AstraZeneca R&D, Mölndal, Sweden
Received August 15, 2005
Abstract:
Four of the most well-known, commercially available docking programs, FlexX, GOLD, GLIDE, and ICM, have been examined for their ligand-docking and virtual-screening capabilities. The relative performance of the programs in reproducing the native ligand conformation from starting SMILES strings for 164 high-resolution protein-ligand complexes is presented and compared. Applying only the native scoring functions, the latest versions of these four docking programs were also used to conduct virtual screening for 12 protein targets of therapeutic interest, involving both publicly available structures and AstraZeneca in-house structures. The capability of the four programs to correctly rank-order target-specific active compounds over alternative binders and nonbinders (decoys plus randomly selected compounds) and thereby enrich a small subset of a screening library is compared. Enrichments from the virtual-screening experiments are contrasted with those obtained with alternative 3D shape-matching and 2D similarity database-search methods.
With the advent of high-performance and low-cost computing systems, exemplified by enterprise grid-based networks and large Linux farms, the past decade has been witness to a major change in the practice of molecular modeling in the pharmaceutical industry, particularly in the resources available to the computational chemist.1 As a result, computational methods are being increasingly used in various stages of the drug-discovery process.2 Coupled with a rapidly rising number of protein structures, structure-based drug design, driven by molecular docking and binding prediction, has been undergoing somewhat of a renaissance.3
Molecular-docking methodologies ultimately seek to predict (or often retrospectively reproduce) the best mode by which a given compound will fit into a binding site of a macromolecular target. Docking, as a result, usually involves two independent steps: (1) the sampling of the ligand's positional, conformational, and configurational space to predict the ligand's pose within the binding site of the receptor and (2) the scoring of the ligand's pose such that the ranking typically is an arbitrary reflection of how well a ligand is expected to bind to its cognate receptor. The docking dilemma thus requires an efficient sampling and searching procedure that is a necessary prerequisite to accomplish point 1 and an accurate scoring function that will correctly assign the bound ligand a priority score that will successfully achieve point 2.
The re-emergence of such in-silico-based screening methods is of practical importance for lead-compound generation in drug discovery.4 Molecular-docking programs coupled with suitable scoring functions are now very much established as the necessary tools that enable computational chemists to rapidly screen large chemical databases and thereby identify promising candidate compounds for further experimental processing.5 A number of docking programs such as DOCK,6 FlexX,7 GOLD,8 AutoDock,9 GLIDE,10 QXP,11 and ICM12 have been developed for just this purpose. Consequently, molecular docking has caught the attention of many pharmaceutical and biotechnology companies eager to discover novel chemical entities, and this has culminated in several well-documented comparative benchmarks on the relative performance of one docking code versus another, including various combinations of those noted above.13 Such studies are very much of interest to us, but as has been pointed out recently,14 and as we are only too aware, because of the many control parameters and other variables that influence the results of each docking code, docking comparisons are extremely difficult to do without inadvertently (dis)favoring the performance of one code over another. Regardless of how objective or careful the authors have strived to be, the results of such comparisons can often be misleading or not generally applicable to the problem at hand.
Given the increased use of molecular-docking approaches and the growth in the number of modeling toolsets available with docking capability, a comprehensive evaluation of a number of docking programs over a large and highly curated data set containing several protein classes of therapeutic interest is clearly useful. In this sense, AstraZeneca is no different from the other pharmaceutical and biotechnology companies that have benchmarked docking-software performance, in that we have also performed evaluations which allow us to gauge the current state of the art. From our viewpoint, such a study provides invaluable information which enables us to devise improved protocols for small-molecule docking and subsequent virtual-screening campaigns, while also serving to highlight areas for improvement in the next generation of docking programs and scoring functions.
From the perspective of hit or lead identification, a more significant aspect is the relative performance of different docking programs as efficient and effective virtual-screening engines. Here, the ability to separate a small subset of active compounds for a given protein target from a sufficiently large set of randomly selected compounds is of paramount importance, as opposed to producing a correct correlation between the calculated and experimental binding affinities. Of course, the most essential component of being able to score ligands appropriately and reproduce credible enrichment statistics for a specific target is to first be able to predict the binding mode of a molecule correctly. A good rule of thumb is that, if the ligand is not docked in the correct conformation, it is very unlikely that the calculated score and associated priority ranking a compound receives is of any significance. However, exceptions to this rule can arise, particularly when ligand poses are fortuitously close-lying in energy. In addition, correctly predicting ligand binding will assist in the subsequent selection and optimization of compounds.
In this article, we report the results of an extensive study of the docking accuracy of four programs, FlexX, GLIDE, GOLD, and ICM. As noted previously, a concern with all published docking evaluations is that they are inherently difficult to do without accidentally biasing the performance of one code over another. In this respect, we have endeavored to be particularly vigilant in our approach and build on the experience of former docking evaluations. To be sufficiently distinctive from all other previously related works, we have tried to ensure that this study both is cognizant of and emphasizes the problematic issues associated with docking comparisons. It is quite likely that the list of topics that we cover is not all-encompassing, but we hope that enough of the major issues that affect the outcome of a comparative evaluation are exposed. Points worthy of note and possible areas of concern are highlighted throughout the text in italics.
All four programs were used to dock random conformers of ligands (starting from SMILES strings, which is a realistic unbiased representation of how compounds are stored in AstraZeneca databases) into the binding site of a receptor (164 in total). The root-mean-square deviation (RMSD) (for the heavy atoms only) between the docked conformers (rank #1) and experimental X-ray structures were then calculated and compared using a stricter measure of success than has been used in the majority of the previous evaluations. The objective of this docking exercise was very simply to assess the ability of each program to reproduce the conformation of the native ligand over a wide-ranging data set of approximately 164 X-ray crystal structures. By defining the ligand as a SMILES string and running each program at the default settings by following the instructions recommended in the various user's guides, we acquire a sense of the general performance capabilities of each program. This obviously does not preclude the possibility that a particular program will perform significantly better in the hands of an expert.
All four programs were then used to dock a carefully designed database (compound structures stored in SMILES format) of randomly selected drug-like compounds plus known receptor-specific active ligands of varying activity against a broad range (12 in total) of pharmaceutically relevant protein targets. As an example of the type of challenge we were setting, for one target in particular, 17 actives were to be extracted from a much larger database of ~40 000 compounds. This level of acute discriminatory capability by a scoring function is very much representative of the type of performance we are looking to implement in everyday discovery research. Macromolecular targets were carefully chosen to represent a range of protein classes having substantially different active site topologies and characteristics. For each target examined, at least one protein structure with a cocrystallized ligand was available in the public domain. The recovery rate was then used to calculate an enrichment factor on a per-target basis as the key measure of how well a particular docking program performed in identifying the known target-specific active compounds from the randomly selected drug-like compounds.
Through this kind of extensive evaluation and comparison, we aim to develop an improved understanding of the relative merits and shortcomings of the docking programs available to us for present-day use in AstraZeneca discovery-research projects.
In this section, we present a detailed description of the procedure used to select and prepare protein receptors and associated ligands for the binding mode prediction and enrichment studies. The methodology associated with the various docking programs is also described briefly.
Target Selection for Binding-Mode Assessment. A total of 164 protein-ligand complexes were selected from the Protein Data Bank (PDB)15 according to the following criteria:
General Features
 Noncovalent binding between ligand and protein
Noncovalent binding between ligand and protein
Crystallographic resolution around 3.0 Å or better
Ligand Features
Molecular weight between 150 and 800 Da
From 1 to 16 rotatable bonds, that is, varied flexibility
of receptor-bound ligands
drug/lead/nonlead like
structurally diverse
Protein Features
Multiple structural motifs (wide spectrum of receptor
families)
Diversity within classes
Metal present in some of the binding pockets
Range of active site topologies and water accessibility
Activities of bound ligands ranging from low micromolar
to nanomolar
Relevant for drug discovery (for the most part)
Our receptor test set incorporates several well-known protein structural motifs (kinases, proteases, lipases, transferases, isomerases, phosphatases, oxidoreductases, nuclear receptors, heme-containing proteins, a GPCR) and ligands (sugars, macrocycles, and peptidomimetics). Receptors were also chosen with more than one bound ligand per crystal structure, to explore the ability of the docking programs to handle various conformations of the same receptor and eliminate potential failures due to protein rearrangement upon ligand binding (induced fit).16 To ensure diversity in the test set, a number of lesser-quality structures were also included. Proteases and kinases are the most widely represented families in the test set, a statistic which merely illustrates that these two protein classes have long been, and still are, to some extent, the focus of modern structure-based drug-discovery programs. The thresholds for molecular weight and number of rotatable bonds of the ligands were set to reflect a wide distribution of potential molecules, covering both drug-like and nondrug-like structures. In summary, we believe that our data set of ~164 complexes is broad and provides a challenging test of binding mode prediction capability.17
Receptor Preparation For Binding-Mode Assessment. Generally, for the 164 protein targets, if a cofactor was present at the binding site, its bond order and protonation state were inspected and corrected if required. When relevant, metal ions at the binding site were preserved. With only a few tightly bound exceptions (PDB ids: 1dwd, 1lna, and 4phr), all the crystallographic waters were deleted from the binding pockets. After removal of the ligand, solvent, and cofactor (when the latter two were not intrinsic parts of the binding site), additional domains not involved in ligand binding, stabilizing counterions, and other extraneous small molecules far from the active site were also removed. Residues at the binding site of each receptor were then visually inspected, hydrogens were added along with missing heavy atoms and partial charges, corrections were made to the orientations of hydroxyl groups and disulfide bonds, and the tautomeric states of histidine residues and the protonation states of basic and acidic residues were adjusted to be the dominant species at pH 7.0. Hydrogen atoms were added in Sybyl 6.5,18 and a constrained minimization was then used to optimize the hydrogen positions. The end product of this process was a clean receptor Sybyl mol2 file for docking studies.
In the process of receptor preparation, it would appear wrong to us to mix different assumptions between different docking programs. By that, we mean that, potentially, it could be a fatal flaw in our approach to presume that receptor preparation by one program will be eminently transferable to and compatible with another program, especially when some programs have their own receptor preparation protocols governing details such as atom-type assignment. Cognizant of this fact that differences in atom-type assignment may ultimately affect the accuracy of the grid potentials calculated for both GLIDE and ICM, and in accord with the general criteria outlined above, protein receptors for GLIDE were prepared from the PDB structures with the protein preparation procedure within the Schrödinger program Maestro.19 For ICM, the raw PDB structure was converted to an ICM object in order to impose the internal coordinate tree on the original Cartesian coordinates of the protein receptor. The conversion process to internal coordinate space is also in accord with the procedure described above and yields a clean and healthy receptor for docking.20
Ligand Preparation for Binding-Mode Assessment. The X-ray coordinates of the ligands were extracted from each of the 164 protein receptors. Each ligand was examined for bond order and protonation state, and written out as a three-dimensional "reference ligand" Sybyl mol2 file and as a predocking Sybyl mol2 file for further processing. The predocking ligand Sybyl mol2 file was converted into SMILES format with an in-house program called SDFilter with the correct stereochemistry (to ensure production of the correct invertomer or ring conformation upon conversion to 3D). As randomization of the starting geometry and position of the ligand are both important, each SMILES string was then converted to fresh and, therefore, unbiased (in terms of 3D conformation and Cartesian position with respect to the original position and coordinates of the native ligand) 3D conformations with Corina,21 further minimized in Sybyl, and then saved as an independent mol2 file ready for docking. The ionization states of the ligands we were attempting to dock were also of particular concern; thus, all carboxylic acids were deprotonated, tertiary amines were positively charged, phosphonates were partially deprotonated, and guanidiniums were positively charged. Pipeline Pilot version 4.522 was used to calculate several molecular descriptors [molecular weight (MW), number of rotatable bonds (NRots), polar surface area (PSA), log D, volume, surface area, etc.] to aid the overall analysis of physicochemical diversity within the ligand set.
Targets for enrichment studies. A selection of 12 receptor targets was used in this part of the study. A total of 8 of the 12 target structures were selected from the AstraZeneca in-house structure collection, comprising nNOS (neuronal nitric oxide synthase, 1.95 Å), CPB (carboxypeptidase, 2.0 Å), HPMurl (glutamate racemase, 2.2 Å), GSK3b (kinase, 2.8 Å), Factor Xa (serine protease, 2.3 Å), P38 (map kinase, 2.0 Å), JNK3 (kinase, 2.2 Å), and PTP1B (phosphatase, 1.8 Å). The other four structures were extracted from the protein data bank and consisted of thrombin (serine protease, PDB id: 1dwd, 3.0 Å), COX2 (cyclooxygenase, PDB id: 1cx2, 3.0 Å), ER (estrogen receptor, PDB id: 1err, 2.6 Å), and sPLA2 (lipase, PDB id: 1db4, 2.2 Å). These 12 targets were prepared for virtual screening in accord with the procedure described above for the binding-mode prediction receptors.
Docking Library for Enrichment Studies. A key issue when evaluating enrichment rates is the number and nature of the active compounds that are included in the test set. In this sense, we wish to make clear the distinction between receptor-specific active compounds, other receptor-specific actives, decoys, and randomly selected compounds.
For the 12 pharmaceutically relevant protein targets listed above, a total set of 2743 active ligands was compiled on the basis of the public literature23 and the AstraZeneca high-throughput screening database. The 2734 active ligands were broken down on a target-by-target basis into receptor-specific active compounds according to the following distribution: nNOS (263 ligands), CPB (74 ligands), HPMurl (154 ligands), GSK3b (576 ligands), factor Xa (81 ligands), P38 (26 ligands), JNK3 (537 ligands), PTP1B (622 ligands), thrombin (125 ligands), COX2 (124 ligands), ER (53 ligands), and sPLA2 (17 ligands). Regardless of the receptor target under investigation, all actives were included in the database during a virtual screen. The potencies of the total set of 2743 active ligands were randomly distributed among the 12 targets, ranging from low micromolar to nanomolar affinity, and in principle, as the former are ostensibly more difficult to rank-order correctly than the latter, they are perhaps more representative of the real-world challenge of finding lead compounds in discovery research. In the most extreme cases, sPLA2, FXa, and thrombin, for example, the target-specific actives display some congeneric features and, therefore, provide the most difficult examination of discriminatory capability. All structures were stored in SMILES format.
A subset of commercially available chemicals was prepared by randomly selecting approximately 20 000 compounds from a larger data set containing over 900 000 compound structures. These compounds were also selected to satisfy the following criteria:
General Features
Molecular weight between 150 and 750 Da
log D value between -6 and +6
Number of rotatable bonds less than 7
At least one polar atom (N, O, S, or P)
No reactive functionalities as defined by AstraZeneca
chemists
Actives, decoys (compounds that were similar to the active compounds in every respect except for activity), and random selections (compounds that bear little resemblance to the active ligands) were selected with a similar distribution of molecular weight, to minimize the well-known tendency of a scoring function to favor larger molecules (see below). To ensure that discrimination by a scoring function was a real challenge, the selection of decoys was biased toward drug-like molecules using filters for functional groups and cutoffs for both molecular weight and the number of rotatable bonds. By combining the randomly selected subset with the set of 2743 receptor-specific ligands and removing duplicate structures, we obtained a set of 22 743 compounds ready for further computational processing and docking studies. This SMILES library was then converted to 3D coordinates with Corina, and all possible tautomers and protonation states were enumerated for each compound, yielding approximately 40 000 compounds in total. Each structure was then saved as an independent entry in a Sybyl multi-mol2 file.
Molecular Properties Distribution of Actives and Randomly Selected Compounds. Before running the virtual screening experiments, molecular properties, such as log D, MW, number of rotatable bonds (NRot), PSA, and number of hydrogen bond donors and acceptors (HBD, HBA), were calculated for the target-specific actives and the randomly selected compounds in the screening database. This was done as a cautionary check to examine if any obvious systematic differences in structures and properties existed between the known active ligand set and the randomly selected compounds, to minimize the chances of introducing any potential bias in favor of the former. The results are shown in Figure 1. As drug-like rules were used in the initial selection of the randomly selected compounds, in all the property distribution figures over the ranges indicated, there were no clear differences between known actives and randomly selected compounds. We are, therefore, confident that the set of receptor-specific active compounds and randomly selected compounds are sufficiently alike that they provide an exacting test of the discriminatory capability of the four programs.
 | Figure 1 Physicochemical profile of the target-specific active (blue) and randomly selected (yellow) sets of compounds. |
3D Shape Matching and 2D Similarity Searching. For the enrichment studies by 3D shape matching and searching by 2D similarity, one X-ray crystallography structure was chosen for each target (see Chart 1 for public structures). The bound ligand conformer was extracted and used as a template for 3D shape matching. The influence of the template ligand's conformation on enrichment was also studied. The X-ray conformation of each template structure was minimized in Sybyl with the Merck Molecular Force Field (MMFF),24 and the minimized conformation was used as a template in a 3D shape search. For the shape-matching experiments, no charge or pharmacophore-like feature mappings were considered. The 2D formula of the bound ligand was also used as a query for a 2D similarity fingerprint search. The scores for 3D shape matching and the 2D similarity between the library compounds and the template molecule were then calculated and incorporated into the enrichment study. 3D shape matching was performed using ROCS 2.0,25 and for each structure in the screening library, a maximum of 100 conformers were generated. ISIS/Base 2.326 was used to undertake the 2D similarity search.
 | Chart 1. Structures Used in Shape Matching and Similarity Searching |
Docking Programs. The latest versions of each code available to us at the time, FlexX 1.10, GOLD 2.2, GLIDE 3.5, and ICM 3.2.01a, were used for the docking studies. A brief description is given below for each of the docking programs. For more details, the reader is referred to the original references and user's guides of each code. Binding-mode prediction experiments were carried out on a dual processor (Intel Xeron) Linux workstation, each processor having a clock speed of 2.8 GHz. Using an in-house program called Match3D, which takes into account local symmetry in all cases, the accuracy of each docking prediction was ascertained on the basis of the RMSD between the coordinates of the heavy atoms of the ligand in the top docking pose only (ranked #1) and those in the crystal structure. Visual inspection was also used to verify the docking pose in each case. For the enrichment test, all calculations were performed on a Linux farm consisting of 100 1.0 GHz Intel Pentium III processors.
FlexX 1.10. FlexX is one of the most used incremental fragment-based docking programs inspired by the Leach and Kuntz algorithm.27 In this algorithm, a set of preferred torsional angles (up to 12) extracted from the Cambridge Structural Database is assigned.28 To map the fragment on the protein active site, the following interaction types are taken into account: entropy, hydrogen bonds, metal acceptor, aromatic ring, methyl, and amide.29 The most challenging part of this algorithm is the placement of the base fragment. To be successful, the base should have some putative interactions such as hydrogen bond donor, hydrogen bond acceptor, or interaction with an aryl group. The ligand interaction centers are then mapped on the reversed protein interaction centers, and the best scores are retained.
GOLD 2.2. GOLD is based on a genetic algorithm. The
ligand's state is encoded by a chromosome,30 representing
its conformation and hydrogen bonding. The conformation
of the ligand is represented by a binary string, in which every
byte encodes for one torsional angle. Each torsion is allowed
to vary between -180 and +180 in step sizes of 1.4 Å.
Two integer strings encode mappings suggesting possible
hydrogen bonds between the protein and the ligand. The first
of these strings encodes a mapping of acceptors in the ligand
to the donor atoms in the protein. The second string encodes
a mapping of donor hydrogens in the ligand to acceptor
atoms in the protein. On decoding a chromosome, GOLD
utilizes least-squares fitting to form as many of these
hydrogen bonds as possible. In the evolutionary development
of the ligand conformations, the program employs an island
model, in which several subpopulations of chromosomes are
created at the beginning instead of one large population. The
genetic operations include the migration of individual
chromosomes between the subpopulations, crossover, and
mutation. To preserve diversity within the population, GOLD
employs a niching technique. If there are more than a
specified number of individuals in the niche, then the new
individual replaces the worst member of the niche rather than
the worst member of the total population. Two individuals
share the same niche if the RMSD between their donor and
acceptor coordinates is less than 1.0 Å. The fitness of a new
individual is assessed using a scoring function that includes
energy terms accounting for hydrogen bonding, short-ranged
van der Waals interactions between the ligand and the
protein, and the ligand internal energy. The latter is a sum
of ligand steric and torsional energies.
and +180 in step sizes of 1.4 Å.
Two integer strings encode mappings suggesting possible
hydrogen bonds between the protein and the ligand. The first
of these strings encodes a mapping of acceptors in the ligand
to the donor atoms in the protein. The second string encodes
a mapping of donor hydrogens in the ligand to acceptor
atoms in the protein. On decoding a chromosome, GOLD
utilizes least-squares fitting to form as many of these
hydrogen bonds as possible. In the evolutionary development
of the ligand conformations, the program employs an island
model, in which several subpopulations of chromosomes are
created at the beginning instead of one large population. The
genetic operations include the migration of individual
chromosomes between the subpopulations, crossover, and
mutation. To preserve diversity within the population, GOLD
employs a niching technique. If there are more than a
specified number of individuals in the niche, then the new
individual replaces the worst member of the niche rather than
the worst member of the total population. Two individuals
share the same niche if the RMSD between their donor and
acceptor coordinates is less than 1.0 Å. The fitness of a new
individual is assessed using a scoring function that includes
energy terms accounting for hydrogen bonding, short-ranged
van der Waals interactions between the ligand and the
protein, and the ligand internal energy. The latter is a sum
of ligand steric and torsional energies.
GLIDE 3.5. The GLIDE algorithm approximates a systematic search of positions, orientations, and conformations of the ligand in the protein-binding pocket via a series of hierarchical filters. The shape and properties of the receptor are represented on a grid by several different sets of fields that provide a progressively more accurate scoring of the ligand pose. The fields are computed prior to docking. The binding site is defined by a rectangular box confining the translations of the center of mass of the ligand. A set of initial ligand conformations is generated through an exhaustive search of the torsional minima, and the conformers are clustered in a combinatorial fashion. Each cluster, characterized by a common conformation of the core and an exhaustive set of side-chain conformations, is docked as a single object in the first stage. The search begins with a rough positioning and scoring phase that significantly narrows the search space and reduces the number of poses to be further considered to a few hundred. In the following stage, the selected poses are minimized on precomputed OPLS-AA van der Waals and electrostatic grids for the receptor. In the final stage, the 5-10 lowest-energy poses obtained in this fashion are subjected to a Monte Carlo procedure in which nearby torsional minima are examined, and the orientation of peripheral groups of the ligand are refined. The minimized poses are then rescored using the GLIDE_Score function, which is a more advanced version of ChemScore31 with force-field-based components and additional terms accounting for solvation and repulsive interactions. The choice of the best pose is made using a model energy score (Emodel) that combines the energy grid score, GLIDE_Score, and the internal strain of the ligand.
ICM 3.2.01a. In the ICM approach, the molecular system is described using internal coordinates as variables. Energy calculations are based on the Empirical Conformational Energy Program for Peptides 332 force-field parameters and MMFF partial charges. In the flexible-ligand-rigid-receptor docking, the receptor field is represented by five potential energy maps: electrostatic, hydrogen bond, hydrophobic, and two van der Waals terms. A global optimization procedure is used to undertake an unbiased, all-atom, flexible docking of the ligand within the rigid binding pocket. This procedure consists of the following steps: (1) a random conformational change of the free variables according to the biased probability Monte Carlo (BPMC) algorithm),33 torsion and rotational angles of the ligand, (2) local energy minimization of the analytical differentiable terms, (3) calculation of the complete energy, including nondifferentiable terms, (4) acceptance or rejection of the total energy on the basis of the Metropolis criterion,34 and (5) allocation of favorable conformations to a conformational stack (history mechanism) that both expels from unwanted minima and promotes the discovery of new minima, followed by a return to step 1. The conformational sampling is based on the BPMC approach,35 which randomly selects a conformation in internal coordinate space and then makes a step to a new, random position, independent of the previous one, but according to a predefined continuous-probability distribution. It has been shown that, after each random step, full local minimization greatly improves the efficiency of the procedure.36 However, because some energy terms might have no derivatives or might be very expensive to compute, a double-energy Monte Carlo minimization scheme circumvents these problems by minimizing the energy with respect to the differentiable terms but calculates the full energy also using the nondifferentiable terms. This double-energy scheme allows for the incorporation of complex energy terms, such as surface-based solvation energy, into the global optimization process.
Binding-Site Definitions. In FlexX, the active site and the interaction surface of the receptor were defined by using the X-ray reference ligand and a 10 Å cutoff distance. In GOLD, the binding site was defined as a spherical region of 10 Å radius centered on the center of mass of the native ligand. In GLIDE, the binding region of the protein was defined by a 1000 Å3 box centered on the center of mass of the X-ray ligand to confine the center of mass of the docked ligand. In ICM, the atoms delimiting the binding site were selected automatically around the binding envelope by the ICMpocketfinder algorithm,37 such that enough residues were included for the correct protein-ligand interactions to be found. In all cases, default settings were used for all other parameters in accord with the user's guides of each code.
Comparison of Docking Accuracy and Reliability. To assess the prediction of binding modes by the four different programs and to characterize the docking accuracy, several parameters are calculated.
The first parameter is the average RMSD for all the top solutions from each docking program. Here, the top solution refers to the conformation of the docked ligand which is ranked number one (#1) by the native scoring function. To maintain simplicity in our analysis and evaluate the usefulness and true predictive capability of these four packages, we chose to focus only on the RMSD results for the top-ranked pose. This was because, as evaluators in an industrial chemical laboratory, we were not interested in whether the four programs could reproduce the experimental binding mode in the top 100 poses; they can for a large majority of cases, and of that we are certain. We were only interested in what they ranked as their #1 pose and whether we could repeatedly rely on this as an indicator of the binding mode. The second parameter is the RMSD of each docking solution. If a docked solution has a heavy-atom RMSD lower than or equal to 2.0 Å, it is regarded as a successful solution. The value 2.0 Å may be interpreted as a rather generous measure of docking success, so we have also enforced a more rigorous measure of docking accuracy by setting the threshold to 1.0 Å rather than the habitually used 2.0 Å.38 We are of the opinion that 1.0 Å is a more meaningful assessment of the type of accuracy we are looking for in every-day small-molecule docking. The final important metric is the success rate, which is the percentage of successful solutions coming from the top solutions at the two different RMSD thresholds.
The results of the binding mode assessment for the top
solutions produced from each docking program's native
scoring function are given in Figure 2 and Table 1 . Generally,
from the scatter plot of the RMSD against the number of
compounds docked, all four docking programs perform
reasonably well against a wide range of targets. ICM and
GLIDE produce the lowest average RMSD matching with
the native ligands, 1.08 and 2.37 Å, respectively, while
GOLD and FlexX fared worse with RMSDs of 2.80 and 3.98
Å, respectively. At the RMSD cutoff of 2.0 Å depicted by
the green box in each plot, ICM and GLIDE performed well
in that they classified 149/164 and 104/164 compounds
correctly within this threshold, giving success rates of 91
and 63%, respectively. GOLD (91 from 164, 55%) also
performed reasonably, while FlexX (70 from 164, 42%)
performed less well. At the more stringent RMSD cutoff of
1.0 Å, denoted by the magenta box in each plot, ICM and
GLIDE again performed well, correctly docking 93/164 and
81/164, leading to success rates of 57 and 49%, respectively.
GOLD was successful in 64 out of 164 cases for a success
rate of 39%, and FlexX was successful 26% (42 from 164)
of the time. Even for the better performing programs, the
docking success rate was reduced considerably at the more
rigorous threshold.
. Generally,
from the scatter plot of the RMSD against the number of
compounds docked, all four docking programs perform
reasonably well against a wide range of targets. ICM and
GLIDE produce the lowest average RMSD matching with
the native ligands, 1.08 and 2.37 Å, respectively, while
GOLD and FlexX fared worse with RMSDs of 2.80 and 3.98
Å, respectively. At the RMSD cutoff of 2.0 Å depicted by
the green box in each plot, ICM and GLIDE performed well
in that they classified 149/164 and 104/164 compounds
correctly within this threshold, giving success rates of 91
and 63%, respectively. GOLD (91 from 164, 55%) also
performed reasonably, while FlexX (70 from 164, 42%)
performed less well. At the more stringent RMSD cutoff of
1.0 Å, denoted by the magenta box in each plot, ICM and
GLIDE again performed well, correctly docking 93/164 and
81/164, leading to success rates of 57 and 49%, respectively.
GOLD was successful in 64 out of 164 cases for a success
rate of 39%, and FlexX was successful 26% (42 from 164)
of the time. Even for the better performing programs, the
docking success rate was reduced considerably at the more
rigorous threshold.
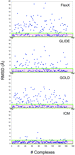 | Figure 2 Comparison of RMSD for rank #1 solutions for ~164 protein-ligand complexes redocked from SMILES. The colored boxes signify the number of docking solutions within a given RMSD threshold level. Colors: green, within 2.0 Å; magenta, within 1.0 Å. |
While the lack of accuracy at the higher RMSD threshold may appear discouraging, it is, of course, instructive to remember that docking-based virtual screening is presently an approximate and, therefore, error-prone science. Even with this caveat, useful information can still be obtained from docking studies that fail to reproduce the observed binding modes. The ability to scope out less-correct but alternative possibilities certainly has valuable implications during the overall drug-design process, where unexpected binding modes can trigger new ideas and potentially lead to improved compounds.
A clear example of misdocking can be seen in the complex between chorismate mutase and prephenate (PDB id: 1com), which displays a small, polar ligand bound to a pocket which is mainly hydrophilic in nature. A number of electrostatic interactions stabilize the complex. The best pose predicted by all the programs shows a flat-ringed structure compared to the peculiar bath-tub-bend conformation observed in the X-ray structure. Clearly, this is a good example of a ligand-ring system which adopts a peculiar conformation in the protein which is not regenerated in the redocking experiment, even though the ring conformation was specified correctly prior to and during 3D conversion. Hence, the likelihood of it ever being docked correctly is small. Although the hydrogen bond between the ligand's hydroxyl group and the protein is retained, the remainder of the molecule is predicted to establish an alternative pattern of salt bridges and hydrogen bonds. As a result, the internal energy of the docked ligand is more favorable than that in the original X-ray structure. Accordingly, the four programs top-scored such a conformation.
In flexible-ligand docking, the size and flexibility of a ligand is known to have a major effect on the docking accuracy. We thus examined this effect by classifying the docking results into five groups on the basis of the NRots of ~164 ligands; this descriptor is correlated with heavy-atom (non-hydrogen) RMSD in Figure 3. Generally, for all the docking programs, the RMSD increases proportionally as ligands become more flexible. This is a well-known problem in the docking arena that can be traced to inadequate sampling of the conformational space, which increases exponentially with ligand flexibility. Thus, thoroughness of the sampling usually has to be partially sacrificed to keep computing time within reasonable limits. Different algorithms use different methods to circumvent the problem and maximize the efficiency of the conformational sampling. FlexX uses an incremental-construction approach, GOLD is based on a genetic algorithm, a hierarchical systematic search is implemented in GLIDE, while a biased-probability Monte Carlo scheme is at the very foundation of ICM. From Figure 3, we note that both ICM and GLIDE perform well even for some of the most flexible ligands having greater than 10 rotatable bonds, with a large percentage of the docking solutions coming in at under a 2.0 Å RMSD. This suggests that the stochastic search in ICM and the multistage systematic algorithm in GLIDE both explore the conformational space adequately well. Additionally, the binding-mode docking performance as a function of the druggable pocket's shape and volume has also been examined (data not shown). The observed trends are very similar to those presented in Figure 3.
 | Figure 3 Docking RMSD as a function of the number of rotatable bonds of the ligands (NRots). |
Another important finding comes from the regression line for each docking set. An approximate 2-3-fold loss in accuracy can be seen on going from rigid compounds to extremely flexible ligands for GLIDE and GOLD; a factor of 5 is seen for FlexX. ICM performance is somewhat insensitive to NRot, and therefore, increased ligand flexibility would appear to present fewer problems for the stochastic approach implemented in ICM. In the limit of extreme rigidity (NRot = 0), there is less variation in the calculated RMSD among the four docking programs, and all programs dock these compounds well. However, as we tend toward the limit of extreme flexibility (Nrot > 25), it would appear the chances of obtaining a more reliable docking solution are increased with ICM and GLIDE, with the latter losing some of its predictive power. This offers at least a partial explanation for the performance of each docking program against the test set.
The docking results for the four programs are plotted against the resolutions of the 164 protein targets in Figure 4. The protein target set for the redocking study was chosen for historical reasons within AstraZeneca and comprises a number of high- and low-resolution receptors. Even though a more recent higher-quality data set could, in principle, have been compiled, the lesser-quality structures were purposely retained to include diversity and to also enable us to assess if there were any obvious differences in the capabilities of the four docking programs to correctly place ligands into poorer- versus higher-quality structures. Furthermore, it is also important to note that, for many protein targets in drug discovery projects, the 3D structures are solved at medium-to-low resolutions (>2.0Å) because of difficulties during the production of crystals or crystallization procedures employed. Nonetheless, such medium- or low-resolution structures still provide excellent starting points for drug hunting. It is, therefore, of the utmost importance to evaluate how docking programs perform in these cases, to fully assess their contributions in real-life examples. It is clear from the plot that no obvious trends appear in the results, both in the redocking experiments and in the subsequent enrichment studies. In fact, the data are quite illuminating, when one considers that a better-quality data set might be considered to provide a better measure of docking accuracy; Figure 4 clearly shows that, even though we have a large number of receptors having 2 Å resolution or lower, the accuracy of docking does not decrease in accord with the decreasing resolution of the protein. In fact, for some high-resolution structures, docking accuracies are quite poor.
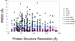 | Figure 4 Docking RMSD as a function of the resolution of protein structures used in the pose prediction test set. |
As several protein families exist in the test set-proteases, kinases, transferases, oxidoreductases, and so forth-the docking results can also be analyzed on the basis of individual target classes. On analysis of the RMSD value with the average rotatable bond number in each protein family, it is worth noting that a greater flexibility of the ligands does not necessarily result in a larger RMSD for some docking programs. For example, in the GLIDE solutions, the RMSD value of aspartic protease inhibitors is lower than that of serine protease inhibitors, while the number of rotatable bonds, and therefore the average flexibility of aspartic protease inhibitors, is larger than in that of serine protease inhibitors. The reverse trend is observed with ICM. Such differences are clearly dependent on the nature of the binding site for the different protein families and also the docking program used.
One final point worthy of note concerns the time taken on average to dock all 164 compounds on a typical desktop computer having a processor speed of 2.8 GHz. As generally the computational speed of each docking code is very relevant for virtual-screening purposes, it is worth mentioning here as part of the comparison. Average calculation speeds for all four docking programs are given in Table 1, with FlexX being the fastest, GOLD the slowest, and ICM and GLIDE representing a compromise between these two extremes. With the latest versions of the codes, the time of docking no longer appears to be a parameter that enables us to discriminate one code over another, as may have been the case for previous slower versions of some of the codes. Given that all the latest versions appear to perform at about the same speed on the basis of the cross-program equivalencies in timings, the more limiting factor would appear to be the accuracy and general reliability of the sampling procedure of the docking code, particularly when a more stringent measure of docking correctness is enforced.
Enrichment Study. The purpose of most general virtual-screening campaigns is to select a subset of a library enriched
in compounds relative to the entire collection and having
the desired activity toward a particular target. When the
percentage of active compounds in the screening set is known
or can be reliably estimated, the success can be quantified
by the enrichment factor. The enrichment factor is defined
as the ratio between the percentage of active compounds in
the selected subset and the percentage in the entire database.
For the purposes of this study, we define the enrichment
factor as
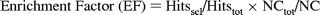
Therefore, when the subsetting level is set at 10%, the theoretical maximum that the enrichment factor can be is 10. In practical virtual screening, however, it is common practice to select a top portion of a library of ranked compounds for further evaluation, but the size of such portions is somewhat arbitrary and, clearly, extremely dependent upon the initial library size. Generally, selections ranging from 0.1% to an upper extreme of 10% of the entire ranking are considered possible, and depending on the value selected, the calculated enrichment factors will differ, as they are themselves dependent upon the fraction of the ranking considered.
The aim of this second part of the evaluation was to examine the performance of the four docking programs as virtual-screening engines. To assess effectiveness, two parameters were calculated to characterize efficiency.
The primary measure of performance we defined as the ability of the docking program to assign priority and rank-order receptor-specific molecules over all of the other molecules in the database. In principle, the specific actives for a target, or a large percentage thereof, should be ranked best. In practice, this may not be achieved, and further analysis may be needed to examine which of the target-specific actives are present in the various upper fractions of the ranked list. Equally important, for a screening method to be of general utility, an acceptable level of performance against a range of different targets is also required, and this is another aspect of our evaluation that we are most interested in. Importantly, this study involves no rescoring of the docking results with alternative scoring functions, that is, consensus scoring. In practice, the rescoring of docking results represents an additional complicating step and a further challenge to the analysis, because meaningful rescoring demands some local reoptimization of the docking poses being rescored according to each of the rescoring functions. Even though the results may, or may not, have improved in some cases, our study was confined to an examination of the performance of the native scoring functions, as they were developed by the authors of the various programs, against a range of targets. A secondary measure of performance, and perhaps one of lesser importance than enrichment, is the screening percentage. We define the screening percentage to be the fraction of the whole library that needs to be screened in order to recoup a certain percentage of known ligands. For the purposes of this study, the screening percentage represents the proportion of the whole library that would need to be screened physically according to the selection such that 80% of the receptor-specific ligands are recovered.
Comparing Performance of Native Scoring Functions.
The general performance of the four docking programs along
with ROCS shape matching and ISIS 2D similarity searching
in the enrichment evaluation is summarized in Figure 5. The
associated data are presented in Table 2.
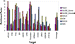 | Figure 5 Enrichment performance of four docking programs, ROCS shape matching, and ISIS 2D similarity searching. For distinction, purple to yellow color shading indicates structure-based docking; blue to cyan denotes ligand-based methods. |
At a subsetting level of 10%, ICM produced the best results overall with an enrichment factor of 6.1 when averaged over all 12 targets. GLIDE combined with its GLIDE_Emodel scoring function produced the second best set of results with an average enrichment of 4.6, which is identical to that obtained from ROCS shape matching. The similar performance of shape matching and docking in these two instances has some practical implications given the tremendous speed advantage of the former over the latter. However, the results from 3D shape matching are functionally dependent on the nature of the template used in the query, as the enrichment factor falls to 4.3 and is noticeably worse when minimized-ligand conformations are used as the template, rather than the X-ray conformation. The implication from this is that the choice of the template conformation for shape matching is very important, and if the pose is predicted correctly, alternative scoring schemes, like ROCS, can be very fast and extremely efficient. The enrichment factor for ISIS 2D similarity searching is 3.5, which is also noticeably worse than shape matching and the ICM and GLIDE dockings. FlexX, combined with FlexXscore, obtained an enrichment factor of 2.2, which is slightly better than the enrichments of 1.7 produced by GOLD and its native scoring function, Goldfitness. This ordering in performance for the latter two docking codes differs from the binding-mode prediction results, where GOLD outperformed FlexX. In all cases, neither docking, shape matching, nor similarity searching obtain a perfect enrichment of 10 over all targets at this level of subsetting. Although ICM and GLIDE come close to achieving perfect enrichment for Factor Xa and thrombin, respectively, only on one occasion for one target, sPLA2, does the GLIDE_Emodel scoring function manage to achieve a perfect 10.
For 7 of the 12 targets, sPLA2, P38, FXa, ER, CPB,
thrombin, and COX2, ICM produces enrichment factors of
 6 and, on average, outperforms all the other methods
examined here in the sense that it produces an acceptable
level of performance across the widest range of targets. On
the basis of the results for this data set, ICM therefore appears
to be the most versatile virtual-screening tool. Using the
Emodel scoring function, GLIDE also performs well for four
of these targets, sPLA2, FXa, nNOS, and thrombin, with
enrichment factors of 6 in each case. The results change
little when GLIDE is combined with its GLIDE_Score
scoring function; in this case, ER scores better than 6 and
nNOS scores worse than 6; hence, ER replaces nNOS in
the above list of four targets. ROCS shape matching also
generated good results for 4 out of 12 targets. Thus, for
sPLA2, FXa, thrombin, and COX2, shape complementarity
between the ligand and the binding site is of premium
importance for binding. It would therefore appear that there
is significant overlap between some targets and their selected
ligands in terms of shape matching. In a somewhat analogous
fashion, for the 2D similarity searching, because the ligands
for FXa and sPLA2 targets share a number of congeneric
features, 2D similarity searching produces a good enrichment
in these two cases. FlexX, with a single enrichment factor
greater than 6, performs well only for thrombin, while GOLD
was unable to produce an enrichment factor 6 for any of
the 12 targets investigated.
6 and, on average, outperforms all the other methods
examined here in the sense that it produces an acceptable
level of performance across the widest range of targets. On
the basis of the results for this data set, ICM therefore appears
to be the most versatile virtual-screening tool. Using the
Emodel scoring function, GLIDE also performs well for four
of these targets, sPLA2, FXa, nNOS, and thrombin, with
enrichment factors of 6 in each case. The results change
little when GLIDE is combined with its GLIDE_Score
scoring function; in this case, ER scores better than 6 and
nNOS scores worse than 6; hence, ER replaces nNOS in
the above list of four targets. ROCS shape matching also
generated good results for 4 out of 12 targets. Thus, for
sPLA2, FXa, thrombin, and COX2, shape complementarity
between the ligand and the binding site is of premium
importance for binding. It would therefore appear that there
is significant overlap between some targets and their selected
ligands in terms of shape matching. In a somewhat analogous
fashion, for the 2D similarity searching, because the ligands
for FXa and sPLA2 targets share a number of congeneric
features, 2D similarity searching produces a good enrichment
in these two cases. FlexX, with a single enrichment factor
greater than 6, performs well only for thrombin, while GOLD
was unable to produce an enrichment factor 6 for any of
the 12 targets investigated.
In practical virtual screening, however, it is a common exercise to select only the top portion of the library of ranked compounds for further evaluation and discard the remainder. The size of the retained portion is logically dependent upon the initial library size and can encompass anywhere from 0.1% to 10% of the entire ranking. From the perspective of aiding our own experimental screening and accelerating lead generation, it would be extremely useful to have a virtual-screening approach that enables us to confidently pick only a handful of the top-ranking compounds for further processing, with some guarantee of a successful hit rate. This becomes ever-more important when library sizes tend toward the order of 1-100 million compounds, as is the case for chemically sensible virtual libraries, for example. Thus, a major purpose of virtual screening is to be able to throw away 99% of the library of compounds virtually screened.
Given that ICM and GLIDE performed well at a 10% level
of subsetting, which is perhaps unrealistic for larger libraries,
we have also compared the performance of these two docking
programs at a more realistic level of 1% subsetting. Data
are shown in Figure 6 and Table 3. Here, a perfect
enrichment factor of 100 is possible only if all the receptor-specific actives are placed in the top 227 scored compounds,
or if receptor-specific actives occupy the top 227 scoring
compounds when there are more than 227 actives to be
recovered. ICM produced the best results overall with an
average enrichment factor of 34. GLIDE combined with its
GLIDE_Score and GLIDE_Emodel scoring functions produced average enrichments of 23 and 21, respectively. For
two targets, P38 and COX2, we note that the GLIDE_Emodel
scoring function does not manage to prioritize any receptor-specific ligands in the top 227 compounds.
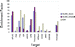 | Figure 6 Enrichment performance for ICM and GLIDE at 1% subsetting. |
As noted previously, the screening percentage is another
measure by which we can further evaluate the performance
of docking programs and their native scoring functions.
Screening percentages are plotted in Figure 7, and data are
shown in Table 4. ROCS shape matching produces the best
results with a screening percentage of 31% but only when
the X-ray ligand conformation is used as the shape-query
template. ROCS's performance deteriorates considerably to
64% when a minimized-ligand conformation is used. Among
the four docking programs, the ICM and GLIDE screening
percentages are comparable at around 45% and represent a
significant improvement over the 63% required by FlexX
and the 85% needed by GOLD.
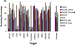 | Figure 7 Screening percentages of four docking programs, ROCS shape matching, and ISIS 2D similarity searching. For distinction, purple to yellow color shading indicates structure-based docking; blue to lilac denotes ligand-based methods. |
Some of these screening percentages may, upon first inspection, appear alarmingly high. On this point, it is worth noting that the recovery beyond 50% or more of the actives can be problematic when one is restricted to flexible-ligand rigid-receptor-based docking. This is because, at high recovery percentage thresholds, such as the 80% we have enforced here, the problem is no longer reduced to a "pure docking ligands that will fit into the binding site and scoring exercise". It is likely that a large number of compounds will, in fact, not fit, and thus, the dilemma becomes one of modeling receptor rearrangement upon ligand binding, that is, induced fit within the binding envelope. As a result, it is not surprising that a large number of compounds are either misdocked and score poorly or not docked at all, and this is reflected in the large percentages of the database needed to be screened to recover 80% of the actives. Nonetheless, for flexible-ligand rigid-receptor-based virtual screening, the results of virtual screening on this data set point unequivocally to the fact that, by using ICM or GLIDE, we are able to screen a much smaller percentage of the library in order to find the same number of active ligands.
The performance of four of the most highly regarded docking programs has been compared. FlexX, GOLD, GLIDE, and ICM were used to redock a data set of 164 ligands into their corresponding receptor binding sites. To gauge the docking accuracy, the non-hydrogen-atom RMSD between the predicted and actual conformers was compared at a generous threshold of 2.0 Å and at a more stringent threshold of 1.0 Å. In this test, ICM, GLIDE, and GOLD achieved respectable performance, with more than 50% of the ligands docked correctly at the more-relaxed threshold; all programs fared poorer when the RMSD was tightened to 1.0 Å, with only ICM and GLIDE correctly docking about half of the ligands within this metric.
To estimate the virtual-screening effectiveness, these four docking programs were also used to conduct screening against 12 protein targets of therapeutic interest, involving both publicly available structures and AstraZeneca in-house structures. The capability to correctly rank-order target-specific active compounds over alternative binders and nonbinders and, thus, enrich a small subset of a screening library was examined. On a target-by-target basis, GLIDE would appear to be the best choice for sPLA2, nNOS, ER, and thrombin; ICM would appear a better choice for FXa, CPB, PTP1B, GSK3B, JNK3, P38, COX2, and HPMurl. When methods other than docking are considered, ROCS shape matching outperforms all four docking methods for COX2 and HPMurl. Also of paramount importance is the performance capability of a scoring function from one target to the next. Taken on average, ICM displays the best enrichment factor of 6.1 at a 10% subsetting, and this is noticeably better than ROCS shape matching (4.6), GLIDE_Emodel (4.6), GLIDE_Score (4.3), ISIS 2D similarity searching (3.5), FlexX (2.2), and GOLD (1.7).
One of the goals of this study was to perform a comprehensive assessment such that it would enable us to implement a set of platform-independent docking and virtual-screening tools that medicinal chemists could apply routinely to as many new targets as possible, prior to experimental high-throughput screening. Therefore, the generality of a program's performance can arguably be taken as a coarse indicator of potential transferability to new targets. From Figures 2-4, given their overall performance in this evaluation, ICM and GLIDE are found to be the most reliable in reproducing the X-ray pose. ICM and GLIDE, therefore, appear to be the primary choices for everyday molecular docking. From Figures 5-7, by using ICM or GLIDE, the chances of obtaining a superior enrichment factor, on average, would appear to be better, and these programs also appear to be excellent choices for virtual screening. But, this we can only confidently say for the data set we have examined here. Certainly, this may not always be the case and we acknowledge that additional tests of virtual screening capability over several different receptor data sets and ligands would be necessary to fully substantiate whether the kind of performance we have observed here is truly generally transferable. Nevertheless, the current benchmark has proven useful in that it enables us to prioritize the selection of docking tools for a number of therapeutically relevant targets.
Conclusions such as those we have stated above, or for that matter, those coming from any other docking-program comparison, are likely subject to several limitations. A small selection is discussed briefly below. It is certainly not our intention to recommend to others a pecking order for the examined docking tools. For our own purposes, we have merely strived to be cognizant of the advantages and disadvantages of the selected tools over a very limited range of targets. As we have had the novice user very much in mind during the evaluation, the results for the four programs were compatible with out-of-the-box settings, which might be of more significance to the medicinal chemist who is interested in experimenting in molecular modeling, rather than the expert modeler. Clearly, for some of the codes, there are many control parameters that influence results, and the benchmarks reported here may, or may not, differ significantly when the programs are run under optimal conditions to achieve peak performance.
One such parameter is the rate at which a compound is docked, that is, the time allocated for sampling in the binding pocket. A number of the programs have different speed settings and so may be classified as either slow or fast depending on the settings chosen. This makes it entirely possible that one program might perform better when fast settings are chosen, but worse when slow settings are enforced and vice versa. It is a common strategy to equalize computing time in an attempt to promote fairness of comparison; we can only conclude that this can be unduly damning to performance in some cases, and we have used the default settings recommended by the authors in each case.
Many of the papers that we have cited earlier in this paper refer to the performance of a program developed by that program's authors with other docking codes. A general observation appears to be that only very rarely are independent workers able to match the docking success rates achieved and published by the vendors of the docking programs. Such differences in performance can, arguably, be ascribed both to a lack of familiarity with the docking code and experience in how to get the best out of the program. For ICM and GLIDE, our observations and the results presented herein are somewhat supportive of the authors' claims of performance, both as docking tools and virtual-screening engines, and this is reassuring.
Being fair to the vendors, it can also be difficult to match the docking success rates achieved and published even by other independent workers. The results of this study are consistent with those of Cheney and Mueller, who reported that, for top-ranking pose prediction accuracy, ICM was 83% reliable compared to GOLD (79%), GLIDE (55%), and FLO (64%).39 This contrasts the study by Perola et al.,13c where GLIDE (61% correctly docked) outperforms both GOLD (48%) and ICM (45%), with the latter performing the poorest of the three programs examined.
The results of the Perola study are clearly at odds with the results that we have obtained here, and at first, we were at a loss to explain the differences. Upon careful examination of their paper, we can only conclude that a possible source of discrepancy that might rationalize the observed difference in ICM performance could arise during the receptor preparation stage. As noted earlier, what would seem wrong to us is to mix different assumptions between different docking programs. To presume that receptor preparation by one program will be eminently transferable to and compatible with another program may be incorrect, especially when programs have their own internally consistent approaches to receptor preparation. In an ideal case, all programs would be compatible with each other in terms of transferability, with universally agreed-upon atom-type definitions and assignments. But, from our experience, this is not the case. Receptor preparation by Macromodel (Schrödinger) is appropriate for GLIDE but not ICM (Molsoft) as some atom-type assignments generated by the former are incompatible and, therefore, unreadable by the latter. Consequently, if any of the critical atoms, which are unreadable by ICM, did happen to be in the binding site, then conceivably this would lead to an inaccurate and, hence, incorrect grid-potential representation of the binding envelope. Docking would therefore take place into a nonsensical description of the binding site for certain targets. Ultimately, this would serve to exaggerate the difference in ICM performance between GLIDE and GOLD for some targets. This is the most plausible explanation for the difference in ICM performance between the Perola study and our own, but clearly, there are potentially many other reasons: versions of the code used [with later releases presumably much improved over earlier ones to corroborate this point; the Vertex study was recently repeated by the same authors using the current version of ICM, and considerable improvements in ICM docking accuracy were observed. The results will be reported separately (Perola, E.; Totrov, M., personal communication)], speeds of the computer processors on which the calculations were performed, the definition of the binding site (this is inherently more difficult to make equal as it can be very subjective. Binding site size equates to search space; the larger the site, the longer and more difficult the search becomes, and an inevitable consequence of large binding site definitions is time spent sampling areas of the protein other than the intended target area), the human element in the study, and so on.
The inability of others to reproduce results in papers such
as those cited and the present one (whether written by
vendors or users) is a serious problem. As a step toward
enabling others to investigate the results of this article without
breaching any level of our own confidentiality, we provide
information concerning known active ligands used in the
enrichment studies. Clearly, we cannot provide information
with regard to proprietary compounds, which were used for
seven receptors, but for the remaining 5 out of the 12 targets,
sPLA2, ER, COX2, P38, and thrombin, active ligands were
compiled from public sources, and a representative example
of each structure is shown in Figure 8. The scores of these
compounds as produced by the four docking programs along
with their rankings relative to the comparison database are
also reported in Table 5. The data presented are consistent
with the overall picture obtained from this evaluation, in that
ICM scores and ranks the active ligands more reliably than
the other three programs. In this way, we hope that by
providing this sort of information, we offer a first step in
the right direction toward enabling these results to be checked
by other workers, for example, with respect to receptor
preparation, or, in cases where receptor structures are
proprietary, to see what happens if a different receptor
conformation is employed.
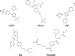 | Figure 8 For 5 out of the 12 targets examined in the virtual screening tests, a publicly available active compound is shown. These structures are associated with the data shown in Table 5. |
As a result of all the possible variables, we are perhaps overly cautious and critical, both toward our own results and toward those of others that have performed comparative docking analyses. With the large number of docking studies now in the literature seemingly playing the performance of one docking program off against another, it is perhaps also likely that the vendors themselves no longer pay too much attention to the results of competitive benchmarking. Reasons for this include those that we have highlighted above, and perhaps others we have not, but it is clear that docking performance can, and does, differ, depending on a number of factors that are often difficult to equalize. Sometimes, in an attempt to promote fairness to all programs by attempting to equalize all parameters, one ends up unintentionally penalizing performance, and this ultimately makes docking comparisons very difficult to do well. For an excellent account of most of the relevant issues, see ref 14.
Even with all these obstacles in mind, docking studies still remain of interest and can be exceptionally useful. Of the published studies to date, this study involves a reasonably large data set for docking-accuracy comparison and covers a wide range of targets for enrichment experiments; in its entirety, the data set is currently one of the most comprehensive that we are aware of. By not optimizing the individual docking protocols, our comparison focused on identifying and highlighting the basic performance factors for each docking program against targets of therapeutic relevance. With the pharmaceutical industry's need for improved lead finding, the results for some docking programs are quite encouraging. Some practically useful insights into the pros and cons, particularly in relation to general transferability and using the docking methods in virtual screening against specific targets, have been obtained. We hope that these findings will be valuable to both academic and industrial colleagues, particularly medicinal chemists, that aspire to use molecular-docking approaches to accelerate lead-compound generation.
The authors thank all colleagues at the AstraZeneca GDECS Structural Chemistry Laboratory, Mölndal. Within computational chemistry, Stefan Schmitt, Ingemar Nilsson, Karin Kolmodin, Peter Varkony, Sorel Muresan, and Jens Sadowski are thanked for providing some practical help and useful discussions. We also express our thanks to Martin Stahl at Hoffman-La Roche AG and Didier Rognan at the University of Strasburg, France, for providing a selecton of ligands and cleaned protein structures.
* To whom correspondence should be addressed. Phone: +46 31 7065285 (H.C.); +46 31 7065735 (T.L.) E-mail: hongming.chen@ astrazeneca.com (H.C.); timothy.lovell@astrazeneca.com (T.L.).
GDECS Chemical Computing, AstraZeneca R&D.
Cancer Discovery, AstraZeneca R&D.
Medicinal Chemistry, AstraZeneca R&D.
2. (a) Kuntz, I. D.; Meng, E. C.; Shoichet, B. K. Structure-based
molecular design. Acc. Chem. Res. 1994, 27, 117-123. (b) Gane, P.
J.; Dean, P. M. Recent advances in structure-based rational drug design.
Curr. Opin. Struct. Biol. 2000, 10, 401-404.
3. (a) Shoichet, B. K.; McGovern, S. L.; Wei, B.; Irwin, J. J. Lead
discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6,
439-446. (b) Abagyan, R.; Totrov, M. High-throughput docking for
lead generation. Curr. Opin. Chem. Biol. 2001, 5, 375-382.
4. Booth, B.; Zemmel, R. Prospects for productivity. Nat. Rev. Drug
Discovery, 2004, 3, 451-456.
5. (a) Bajorath, J. Integration of virtual and high-throughput screening.
Na. Rev. Drug Discovery 2002, 1, 882-894. (b) Lyne, P. D. Structure
based virtual screening: an overview. Drug Discovery Today 2002,
7, 1047-1055.
(c) Good, A. C.; Krystek, S. R.; Mason, J. S. High-throughput and virtual screening: core lead discovery technologies
move towards integration. Drug Discovery Today 2002, 5, S61-S69.
(e) Li, J.;
Lovell, T. The emergence of in silico methodologies. World Pharm.
Frontiers 2005, in press.
6. Makino, S.; Kuntz, I. D. Automated flexible ligand docking method
and its application for database search. J. Comput. Chem. 1997, 18,
1812-1825.
7. Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible
Docking Method using an Incremental Construction Algorithm. J. Mol.
Biol. 1996, 261, 470-489.
8. Jones, G.; Willett, R.; Glen, R.; Leach, A. R.; Taylor, R. Development
and validation of a genetic algorithm for flexible docking. J. Mol.
Biol. 1997, 267, 727-748.
9. Morris, G. M.; Goodsell, D. S.; Halliday, R.; Huey, R.; Hart, W. E.;
Belew, R. K.; Olson, A. J. Automated docking using a Lamarckian
genetic algorithm and an empirical binding free energy function. J.
Comput. Chem. 1998, 19, 1639-1662.
10. Friesner, R. A.; Banks, J. L.; Murphy, R. B.; Halgren, T. A.; Klicic,
J. J.; Mainz, D. T.; Repasky, M. P.; Knoll, E. H.; Shelley, M.; Perry,
J. K.; Shaw, D. E.; Francis, P.; Shenkin, P. S. Glide: A New Approach
for Rapid, Accurate Docking and Scoring. 1. Method and Assessment
of Docking Accuracy. J. Med. Chem. 2004, 47, 1739-1749.
11. McMartin, C.; Bohacek, R. S. QXP: Powerful, rapid computer
algorithms for structure-based drug design. J. Comput.-Aided Mol. Des.
1997, 11, 333-344.
12. Abagyan, R.; Totrov, M.; Kuznetsov, R. ICM - A new method for
protein modeling and design: applications to docking and structure
prediction from the distorted native conformation. J. Comput. Chem.
1994, 15, 488-506.
13. (a) Kroemer, R. T.; Vulpetti, A.; McDonald, J. J.; Rohrer, D. C.;
Trosset, J.-Y.; Giordanetto, F.; Cotesta, S.; McMartin, C.; Kihlen, M.;
Stouten, P. F. W. Assessment of Docking Poses: Interactions-Based
Accuracy Classification (IBAC) versus Crystal Structure Deviations.
J. Chem. Inf. Comput. Sci. 2004, 44, 871-881. 14. Cole, J. C.; Murray, C. W.; Nissink, J. W. M.; Taylor, R. D.; Taylor,
R. Comparing protein-ligand docking programs is difficult. Proteins: Struct., Funct., Bioinf. 2005, 60, 325-332. (b) Cummings, M.
D.; DesJarlais, R. L.; Gibbs, A. C.; Mohan, V.; Jaeger, E. P.
Comparison of Automated Docking Programs as Virtual Screening
Tools. J. Med. Chem. 2005, 48, 962-976.
(c) Perola, E.; Walters, W.
P.; Charifson, P. S. A detailed comparison of current docking and
scoring methods on systems of pharmaceutical relevance. Proteins:
Struct., Funct., Bioinf. 2004, 56, 235-249.
(d) Bissantz, C.; Folkers,
G.; Rognan, D. Protein-based virtual screening of chemical databases.
1. Evaluation of different docking/scoring combinations. J. Med. Chem.
2000, 43, 4759-4767.
(e) Kontoyianni, M.; McClellan, L. M.; Sokol,
G. S. Evaluation of Docking Performance: Comparative Data on
Docking Algorithms. J. Med. Chem. 2004, 47, 558-565.
(f) Wang,
R.; Lu, Y.; Wang, S. Comparative Evaluation of 11 Scoring Functions
for Molecular Docking. J. Med. Chem. 2003, 46, 2287-2303.
(g)
Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative
evaluation of eight docking tools for docking and virtual screening
accuracy. Proteins: Struct., Funct., Bioinf. 2004, 57, 225-242.
(h)
Bursulaya, B. D.; Totrov, M.; Abagyan, R.; Brooks, C. L., III.
Comparative study of several algorithms for flexible ligand docking.
J. Comput.-Aided Mol. Des. 2003, 17, 755-763.
(i) Ferrara, P.;
Gohlke, H.; Price, D. J.; Klebe, G.; Brooks, C. L., III. Assessing
Scoring Functions for Protein-Ligand Interactions. J. Med. Chem.
2004, 47, 3032-3047.
(i) Warren, G. L.; Andrews, C. W.; Capelli,
A.-M.; Clarke, B.; LaLonde, J.; Lambert, M. H.; Lindvall, M.; Nevins,
N.; Semus, S. F.; Senger, S.; Tedesco, G.; Wall, I. D.; Woolven, J.
M.; Peishoff, C. E.; Head, M. S. A critical assessment of docking
programs and scoring functions. J. Med. Chem. 2005, 48, [ASAP].
15. (a) Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.
N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. The Protein Data
Bank. Nucleic Acids Res. 2000, 28, 235-242.
16. Teague, S. J. Implications for protein flexibility for drug discovery.
Nature Rev. Drug Discovery 2003, 2, 527-541.
17. The PDB identification codes of the publicly available proteins studied in this work along with their resolutions in Å are given: 1aaq 2.50, 1abe 1.70, 1abf 1.90, 1acm 2.80, 1acl 2.80, 1acj 2.80, 1add 2.40, 1aha 2.20, 1apu 1.80, 1apt 1.80, 1aqw 1.80, 1aqx 2.00, 1atl 1.80, 1azm 2.00, 1b8y 2.00, 1baf 2.90, 1bbp 2.00, 1bcu 2.00, 1bhx 2.30, 1bji 2.00, 1cbs 1.80, 1cbx 2.00, 1cil 1.60, 1ciz 1.64, 1com 2.20, 1coy 1.80, 1cps 2.25, 1ctt 2.20, 1dbb 2.70, 1dbj 2.70, 1did 2.50, 1die 2.50, 1dmp 2.00, 1dog 2.40, 1dth 2.00, 1dwb 3.16, 1dwd 3.00, 1eap 2.50, 1ebg 2.10, 1eed 2.00, 1ela 1.80, 1elb 2.10, 1elc 1.75, 1epb 2.20, 1epo 2.00, 1ere 3.10, 1err 2.60, 1etr 2.20, 1ett 2.50, 1fax 3.00, 1fkg 2.00, 1fki 2.20, 1frp 2.00, 1glp 1.90, 1glq 1.80, 1hck 1.90, 1hdc 2.20, 1hfc 1.56, 1hfs 1.70, 1hpv 1.90, 1hri 3.00, 1hsl 1.89, 1htf 2.20, 1hvr 1.80, 1hyt 1.70, 1icn 1.74, 1igj 2.50, 1imb 2.20, 1ive 2.40, 1jao 2.40, 1lah 2.06, 1lcp 1.65, 1ldm 2.10, 1lic 1.60, 1lmo 1.80, 1lst 1.80, 1mcr 2.70, 1mdr 2.10, 1mmb 2.10, 1mmq 1.90, 1mnc 2.10, 1mrg 1.80, 1mrk 1.60, 1mtw 1.90, 1mup 2.40, 1nco 1.80, 1nsd 1.80, 1ohj 2.50, 1okl 2.10, 1okm 2.20, 1pbd 2.30, 1phf 1.60, 1ppc 1.80, 1pph 1.90, 1poc 2.00, 1qbt 2.10, 1qbu 1.80, 1rbp 2.00, 1rne 2.40, 1rob 1.60, 1sln 2.27, 1snc 1.65, 1srj 1.80, 1stp 2.60, 1tka 2.70, 1tng 1.80, 1tnh 1.80, 1tnl 1.90, 1tph 1.80, 1tpp 1.40, 1uag 1.95, 1ukz 1.90, 1ulb 2.75, 1usn 1.80, 1uvs 2.80, 1uvt 2.50, 1wap 1.80, 1web 1.50, 1wec 1.50, 1wed 1.50, 1xid 1.70, 1xie 1.70, 1xug 1.50, 1ydr 2.20, 1yds 2.20, 1ydt 2.30, 2ada 2.40, 2cgr 2.20, 2cht 2.20, 2cmd 1.87, 2cpp 1.63, 2ctc 1.40, 2dbl 2.90, 2gbp 1.90, 2h4n 1.90, 2ifb 2.00, 2phh 2.70, 2sim 1.60, 2tmn 1.60, 2tsc 1.97, 2usn 2.20, 2ypi 2.50, 3aah 2.40, 3cla 1.75, 3erd 2.03, 3ert 1.90, 3hvt 2.90, 3ptb 1.70, 3tpi 1.90, 4cts 2.90, 4dfr 1.70, 4fab 2.70, 4phv 2.10, 4tim 2.40, 4tpi 2.20, 5abp 1.80, 5tim 1.83, 5tln 2.30, 6abp 1.67, 6cpa 2.00, 6rnt 1.80, 7tim 1.90, 8atc 2.50, 8gch 1.60.
18. Sybyl, version 6.5; Tripos Inc.: St. Louis, MO.
19. Glide, version 3.5; Schrödinger LLC: New York, 2005.
20. ICM, version 3.2.01a; Molsoft LLC: San Diego, CA, 2004.
21. Gasteiger, J.; Rudolph, C.; Sadowski, J. Automatic generation of 3D
atomic coordinates for organic molecules. Tetrahedron Comput.
Methodol. 1990, 3, 537-5547.
22. Pipeline Pilot, version 4.5; SciTegic: San Diego, CA.
23. Stahl, M.; Rarey, M. Detailed analysis of scoring functions for virtual
screening. J. Med. Chem. 2001, 44, 1035-1042.
24. Halgren, T. A. Merck molecular force field: 1. Basis, form, scope,
parametrization, and performance of MMFF94. J. Comput. Chem.
1995, 17, 490-519.
25. ROCS, version 2.0; Openeye Scientific Software LLC: Santa Fe, New Mexico.
26. ISIS/Base, version 2.3; MDL Information Systems, Inc.: San Leandro, CA.
27. Leach, A. R.; Kuntz, I. D. Conformational analysis of flexible ligands
in macromolecular receptor sites, J. Comput. Chem. 1992, 13, 730-748.
28. Allen, F. H.; Davies, J. E.; Galloy, J. J.; Johnson, O.; Kennard, O.;
Macrae, C. F.; Mitchell, E. M.; Mitchell, G. F.; Smith, J. M.; Watson,
D. G. The development of versions 3 and 4 of the Cambridge Structural
Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187-204.
29. Böhm, H. J. A novel computational tool for automated structure-based
drug design. J. Mol. Recog. 1993, 6, 131-137.
30. Jones, G.; Willett, P.; Glen, R. C. A genetic algorithm for flexible
molecular overlay and pharmacophore elucidation. J. Comput.-Aided
Mol. Des. 1995, 9, 532-549.
31. Eldridge, M.; Murray, C. W.; Auton, T. A.; Paolini, G. V.; Lee, R. P.
Empirical scoring functions: I. The development of a fast empirical
scoring function to estimate the binding affinity of ligands in receptor
complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425-445.
32. Nemethy, G.; Gibson, K. D.; Palmer, K. A.; Yoon, C. N.; Paterlini,
G.; Zagari, A.; Rumsey, S.; Scheraga, H. A. Energy parameters in
polypeptides. 10. Improved geometrical parameters and nonbonded
interactions for use in the ECEPP/3 algorithm, with application to
proline-containing peptides. J. Phys. Chem. 1992, 96, 6472-6484.
33. Abagyan, R. A.; Totrov, M. M. Biased Probability Monte Carlo
Conformational Searches and Electrostatic Calculations For Peptides
and Proteins. J. Mol. Biol. 1994, 235, 983-1002.
34. Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.;
Teller, E. Equation of State Calculations by Fast Computing Machines.
J. Chem. Phys. 1953, 21, 1087-1092.
35. Abagyan, R. A.; Totrov, M. M. Ab Initio Folding of Peptides by the
Optimal-Bias Monte Carlo Minimization Procedure. J. Comput. Phys.
1999, 151, 402-421.
36. (a) Abagyan, R. A.; Argos, P. Optimal Protocol and Trajectory
Visualization For Conformational Searches of Peptides and Proteins.
J. Mol. Biol. 1992, 225, 519-532. (b) Li, Z.; Scheraga, H. A. Monte
Carlo-Minimization Approach to the Multiple-Minima Problem in
Protein Folding. PNAS 1987 84, 6611-6615.
37. (a) An, J.; Totrov, M.; Abagyan, R. Pocketome via comprehensive
identification and classification of ligand binding envelopes. Mol. Cell.
Proteomics 2005, 4, 752-761. (b) An, J.; Totrov, M.; Abagyan, R.
Comprehensive identification of druggable protein ligand binding sites.
Genome Inf. 2005, 15, 31-41.
38. (a) Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FlexX
incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228-241. (b) Gohlke, H.; Hendlich, M.; Klebe, G.
Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337-356.
39. Cheney, D.; Mueller, L. Evaluation of Strategies for Molecular Docking, 226th ACS Meeting, New York, NY, 2003.
|
recovery success rates (# and %) |
|||||||
|
program |
# complexes |
# at 2 Å |
% at 2 Å |
# at 1 Å |
% at 1 Å |
av. RMSD (Å) |
av. time (min) |
|
FlexX |
164 |
70 |
42.6 |
42 |
25.6 |
3.98 |
0.5 |
|
GOLD |
164 |
91 |
55.4 |
64 |
39.0 |
2.80 |
1.4 |
|
GLIDE |
164 |
104 |
63.4 |
81 |
49.3 |
2.37 |
1.0 |
|
ICM |
164 |
149 |
90.8 |
93 |
56.7 |
1.08 |
1.0 |
|
structure-based method |
ligand-based method |
||||||
|
target |
FlexX |
GLIDE_Score |
GLIDE_Emodel |
GOLD |
ICM |
ROCS X-ray |
ISIS |
|
sPLA2 |
1.88 |
8 |
10 |
0 |
7.6 |
7.06 |
7.06 |
|
P38 |
2.08 |
2.4 |
0.8 |
2.13 |
7.3 |
1.54 |
3.08 |
|
FXa |
3.67 |
7.05 |
8.72 |
3.58 |
9.3 |
8.15 |
8.27 |
|
ER |
0.83 |
7.86 |
3.93 |
0.75 |
6.2 |
3.96 |
4.15 |
|
nNOS |
0.11 |
2.43 |
6.65 |
0.08 |
4.7 |
2.16 |
0.34 |
|
CPB |
3.88 |
4.77 |
5.69 |
4.46 |
6.5 |
3.38 |
2.03 |
|
thrombin |
7.5 |
9.03 |
9.84 |
1.44 |
8.8 |
8.4 |
5.6 |
|
HPMurl |
0.54 |
2.33 |
2.6 |
1.05 |
3.9 |
5.69 |
2.09 |
|
COX2 |
0.52 |
3.05 |
1.83 |
3.55 |
7.1 |
8.06 |
2.66 |
|
JNK3 |
1.75 |
2.19 |
1.51 |
1.35 |
5 |
2.92 |
2.81 |
|
GSK3B |
1.92 |
1.26 |
1.74 |
1.16 |
4.7 |
2.94 |
3.09 |
|
PTP1B |
1.37 |
1.43 |
1.64 |
0.93 |
2.3 |
0.64 |
0.77 |
|
average |
2.2 |
4.3 |
4.6 |
1.71 |
6.1 |
4.6 |
3.5 |
|
target |
GLIDE_Score |
GLIDE_Emodel |
ICM |
|
sPLA2 |
53.3 |
66.7 |
64.7 |
|
P38 |
4 |
0 |
15.4 |
|
FXa |
35.9 |
16.7 |
70.4 |
|
ER |
46.4 |
10.7 |
57 |
|
nNOS |
3.8 |
6.1 |
2.2 |
|
CPB |
21.5 |
41.5 |
40.5 |
|
thrombin |
67.7 |
80.6 |
63.2 |
|
HPMurl |
2.7 |
4 |
18.1 |
|
COX2 |
6.1 |
0 |
27.4 |
|
JNK3 |
4 |
1.7 |
7.5 |
|
GSK3B |
1.7 |
2.3 |
3.7 |
|
average |
23 |
21 |
34 |
|
structure-based methodology |
ligand-based methodology |
|||||||
|
target |
FlexX |
GLIDE_Score |
GLIDE_Emodel |
GOLD |
ICM |
ROCS X-ray |
ROCS min |
ISIS |
|
sPLA2 |
55.4 |
9.3 |
4.6 |
94.8 |
55.5 |
41.1 |
76.5 |
58.2 |
|
P38 |
70.4 |
45.8 |
74.1 |
92.2 |
36.3 |
38.7 |
69.3 |
57.3 |
|
FXa |
45.9 |
6.7 |
4.7 |
86.3 |
4.7 |
7.5 |
17.9 |
6.5 |
|
ER |
75.3 |
5.9 |
67 |
97.8 |
53.9 |
42.8 |
88 |
66.5 |
|
nNOS |
82.5 |
58.2 |
15.4 |
85.3 |
60.1 |
33.4 |
73.3 |
82.7 |
|
CPB |
55.4 |
67.5 |
53.4 |
70.6 |
44.1 |
34.1 |
73.2 |
49.2 |
|
thrombin |
14.7 |
3 |
1 |
85.3 |
12.2 |
6.45 |
14.6 |
12.6 |
|
HPMurl |
82.1 |
83.6 |
85.6 |
84.9 |
70.4 |
38.8 |
76.7 |
75.1 |
|
COX2 |
75.3 |
40.9 |
69.3 |
65.4 |
42.5 |
9.4 |
21.7 |
48.9 |
|
JNK3 |
60.7 |
57.8 |
67.6 |
72.9 |
40.7 |
37.8 |
79.5 |
64.6 |
|
GSK3B |
69.1 |
69.2 |
65 |
95.3 |
56.1 |
38.6 |
83.7 |
61.8 |
|
PTP1B |
69.1 |
72.1 |
73.2 |
84.2 |
68.4 |
45.8 |
96.8 |
82.4 |
|
average |
63 |
43 |
48 |
85 |
45 |
31 |
64 |
56 |
|
score (rank #) |
||||
|
target |
FlexX |
GLIDE |
GOLD |
ICM |
|
sPLA2 |
-23.1 (#4511) |
-10.5 (#2) |
31.7 (#24805) |
-54.7 (#1) |
|
P38 |
-32.6 (#318) |
-7.9 (#2338) |
60.0 (#227) |
-37.0 (#74) |
|
ER |
-11.9 (#25665) |
-9.7 (#30) |
13.9 (#32566) |
-65.9 (#1) |
|
thrombin |
-32.6 (#318) |
-8.3 (#37) |
18.1 (#32688) |
-50.1 (#1) |
|
COX2 |
-28.1 (#866) |
-9.1 (#112) |
60.4 (#100) |
-45.6 (#1) |