MolSoft White Paper by Maxim Totrov, Ph.D., CTO, MolSoft LLC - March 6, 2025
Ligand scoring plays a crucial role in virtual screening, a computational technique used to identify potential drug candidates from large compound libraries.
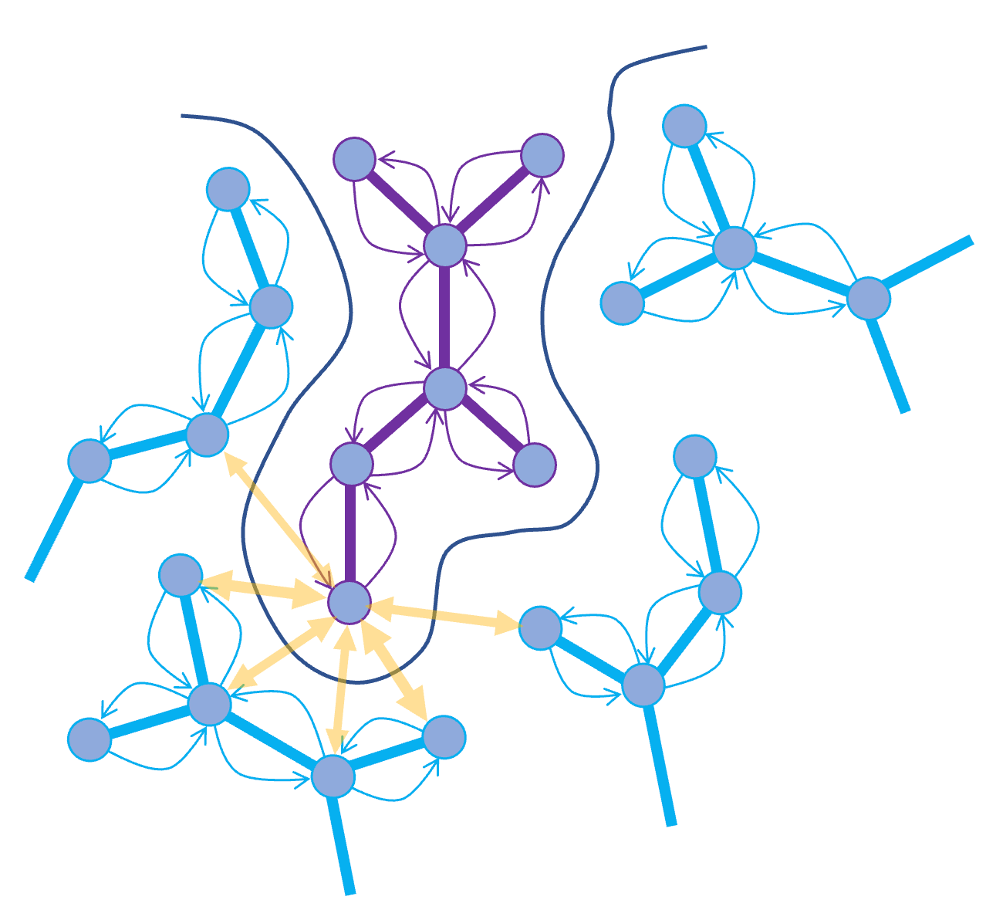 Figure 1. Two types of convolutions in RTCNN
Figure 1. Two types of convolutions in RTCNN
Graph Convolutional Neural Networks (GCNNs) have shown promise in improving ligand scoring by capturing the structural and chemical features of molecules in a graph representation. Traditional ligand scoring methods rely on handcrafted features and simplified physical energy terms that may not fully capture the complex interactions between a ligand and its target protein. GCNNs offer a data-driven approach by learning directly from the molecular graph representation, allowing for more accurate and robust scoring.
The RTCNN graph-convolutional network includes two types of layers (Fig. 1): (1) intramolecular 'topological' 2D chemical connectivity graph atom-to-atom convolutions 1, 2 within ligand and receptor; and (2) intermolecular 3D radial-kernel convolutions between neighboring ligand and receptor atoms, similar to InteractionNet3. The first type of the CNN layers is intended to perceive local chemical environment and deduce interaction properties of different atoms. The second type of CNN layer is designed to perceive the environment (favorable or unfavorable) of each atom in 3D space. Overall NN architecture includes three layers, type 1/type 2/type 1, followed by sum pooling. Input atomic descriptors are: Mendeleevian element descriptor (MED)2, ring membership flag, formal charge, solvent accessibility, number of attached hydrogen atoms, hybridization, and hydrogen bond donor/acceptor flags. Solvent exposed area is included as an additional descriptor for the receptor atoms.
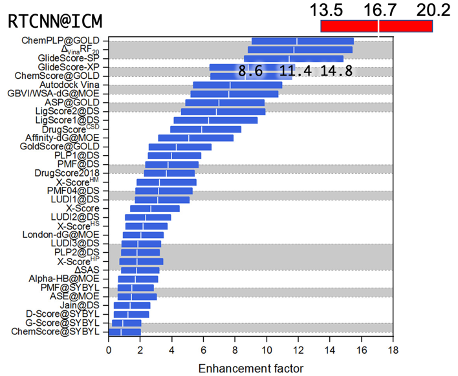 Figure 2. Performance of RTCNN and other scoring functions on CASF 2016 virtual screening benchmark.
Figure 2. Performance of RTCNN and other scoring functions on CASF 2016 virtual screening benchmark.
The network design is intentionally shallow and focuses on local, low-level descriptors which directly relate to the physics of the receptor-ligand interactions, rather than any high level chemical features of the compound or complex global structural features of the binding site, in order to avoid 'memorization' and overtraining. Pose similarity to the native (experimental) structure was used as a loss function. RTCNN is trained on a set of experimental and generated decoy ligand/receptor poses. The training set includes a carefully curated selection of high-quality (<=2.0A) X-ray structures of drug-like (between 200 and 500 MW; N rotatable bonds <= 10; excluding unusual elements, detergents, PEGs, lipids and other common crystallization buffer components ) non-covalent ligand complexes extracted from the PDB. Decoy structures are the non-native-like alternative docking poses produced by icm-dock.Performance of the RTCNN score was evaluated on CASF 2016 benchmark and compared to a range of other scoring functions that were tested in the original benchmark publication
4. Comparison is plotted on Fig. 2; one can observe that RTCNN outperforms other scores by a substantial margin.
References
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P., Molecular graph convolutions: moving beyond fingerprints. J Comput Aided Mol Des 2016, 30, 595-608.
- Raush, E.; Abagyan, R.; Totrov, M., Graph-Convolutional Neural Net Model of the Statistical Torsion Profiles for Small Organic Molecules. J Chem Inf Model 2022, 62, 5896-5906.
- Cho, H.; Lee, E. K.; Choi, I. S., Layer-wise relevance propagation of InteractionNet explains protein-ligand interactions at the atom level. Sci Rep 2020, 10, 21155.
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R., Comparative Assessment of Scoring Functions: The CASF-2016 Update. J Chem Inf Model 2019, 59, 895-913.



