Fast and Accurate Prediction of ADME-TOX Properties
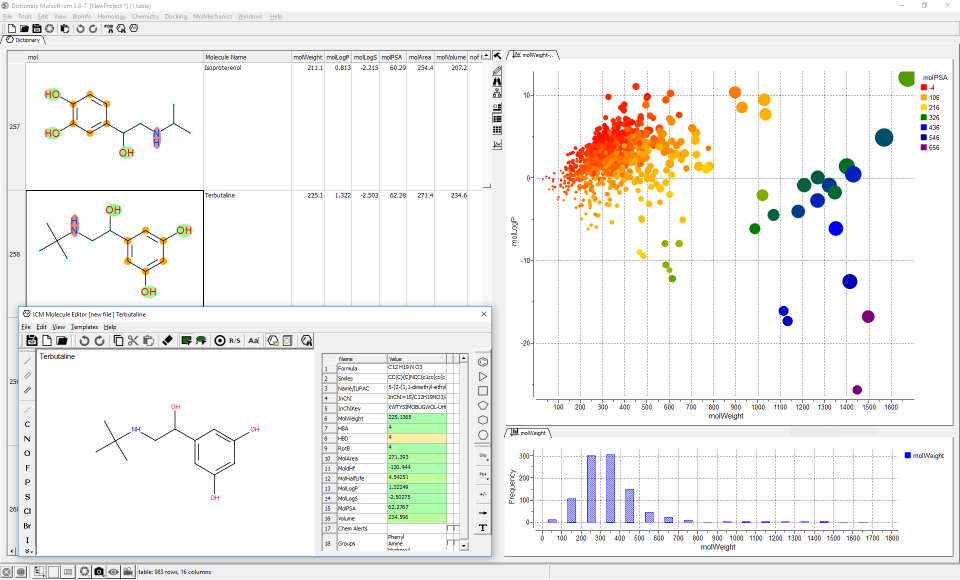
A wide range of ADME properties can be quickly and accurately calculated on large chemical datasets. This tool enables you to flag chemicals that might have poor ADME-TOX properties before experimental testing.
Some of the properties you can predict are:
- MolWeight - Molecular weight from .mol
- MolFormulaChemical - formula,e.g. C2H6O, from .mol
- IupacName - IUPAC nomenclature name from .mol
- MolLogP - Octanol water partition, -Log(C_w/C_oct) from .mol
- MolLogS- Water solubility -Log(C_aggr) from .mol
- MolPSA - Polar surface area from .mol
- MolArea - Total surface area from .mol
- MolVolume- Molecular volume from .mol
- MoldHf - Heats of formation from elements from .mol
- MolhERG - hERG binding prediction
- MolHalfLife - Half life, hours
- MolPAINS" Pan Assay Interference Compounds
- MolCACO2 - Predict CACO-2 Permeability LogP
- MolLD50 - Predict LD50 in mg/kg
- MolPAMPA - Predict PAMPA Permeability
- MolPGPINHIB - Predict P=Glycoportein Inhibition
- MolPGPSUBST - Predict P-Glycoprotein substrate
- DrugLikeness Empirical drug-likeness [-1:+1] from .mol
- Tox Score - Chemical Alert Collected from Chemical supplier and other sources
- Smiles SMILES/SMARTS: string notation of chemical or chemical patterns derived from .mol
- Atom Counts - Atom counts
- Bond Counts - Bond counts
- Topological Descriptors - Toplogical, connectivity and shape indices
- BadGroups - Unwanted or reactive chemical functionality from .mol
- Covalent/Prodrug Groups - Potential chemical groups that can be linked covalently or cleaved in prodrug.
- Nof_Atoms - Number of atoms from .mol
- Nof_Molecules - Number of individual molecules in .mol drawings
- Nof_Fragments - Number of fragments
- Nof_Chirals - Number of chiral centers, R,S,or (RS) from .mol
- Nof_RingsNumber of rings in the SSSR from .mol
- Max_Ring_Size - Largest independent ring size from .mol
- Min_Ring_Size - Smallest independent ring size from .mol
- Max_Fused_Rings - Number of elementary rings in the largest fused ring from .mol
- Nof_RotB - Number of freely rotatable bonds from .mol
- Nof_HBA - Number of hydrogen bond acceptors from .mol
- Nof_HBD - Number of hydrogen bond donors from .mol"
- MolCharge - Calculate total formal charge at given Ph
- pKa of the Most Basic Group - Calculate pKa of most basic group
- pKa of the Most Acidic Group - Calculate pKa of most acidic group
- InChI - Calculate InChi string
- InChIKey - Calclate InChI key string
- PubChem CID = RetrievePubChem CID by structure
- Name or ID from Structure - Convert Structure to Name/CAS o PubChem CID
- ChEMBL Bioactivity - ChEMBL Bioacticity from Structure
- Drug Bank ID - Retrieve Drug Bank ID number
- Structure to Name - Tanslate chemical structure to Name, CAS, DrugBank, ID, ChEMBL ID
- FindChemPatterns - Annotate .mol drawings by found chemical substructure
- 2Dfrom_Smiles - Convert SMILES to 2D .mol chemical drawings
- 2Dfrom_InChi - Convert InChi to 2D .mol chemical drawings
Return to ICM-Chemist Main Page