MolSoft is excited to announce the release of ICM v3.7-2 which is a major new version upgrade with many new features and an updated user interface. You can download the latest version at our support site. Below is a summary of the new features along with some screenshots.
Click here to jump to new feature descriptions.
Screenshots
Screenshots of New Features in Version 3.7-2 - Click to enlarge
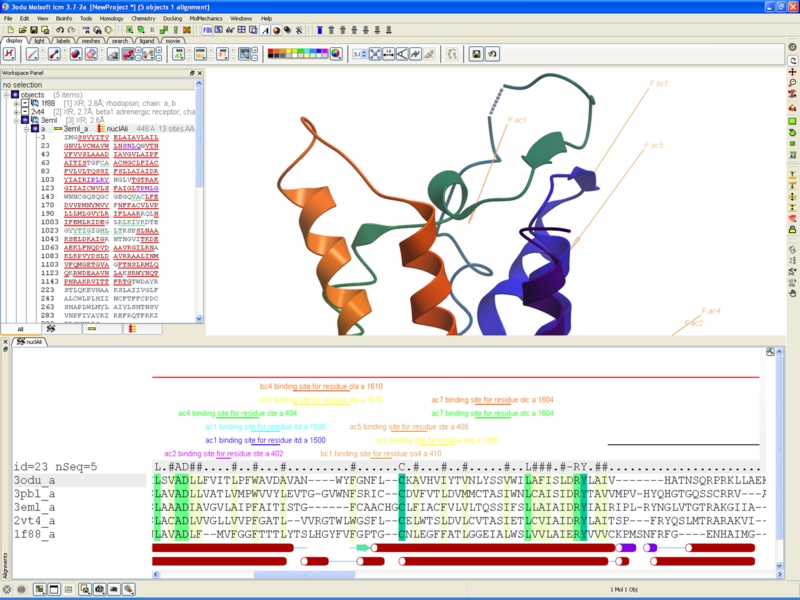
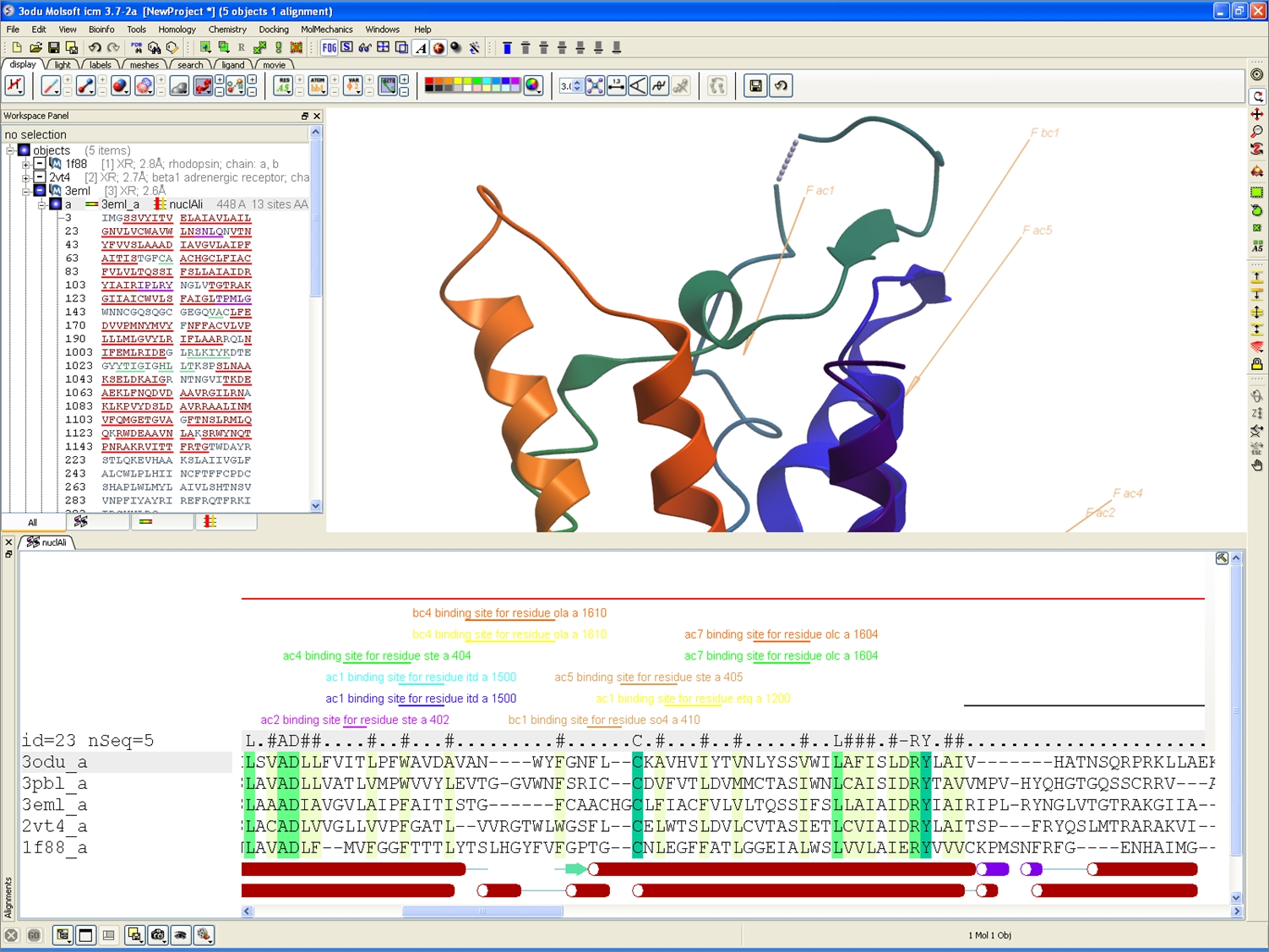
Improved Alignment Annotation
One click display of UniProt (and user defined) annotation on sequence alignments.
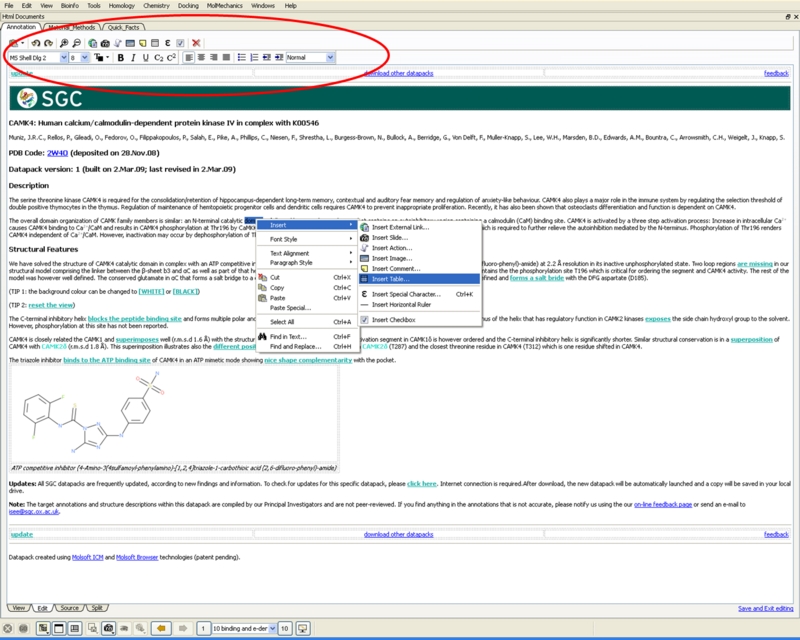
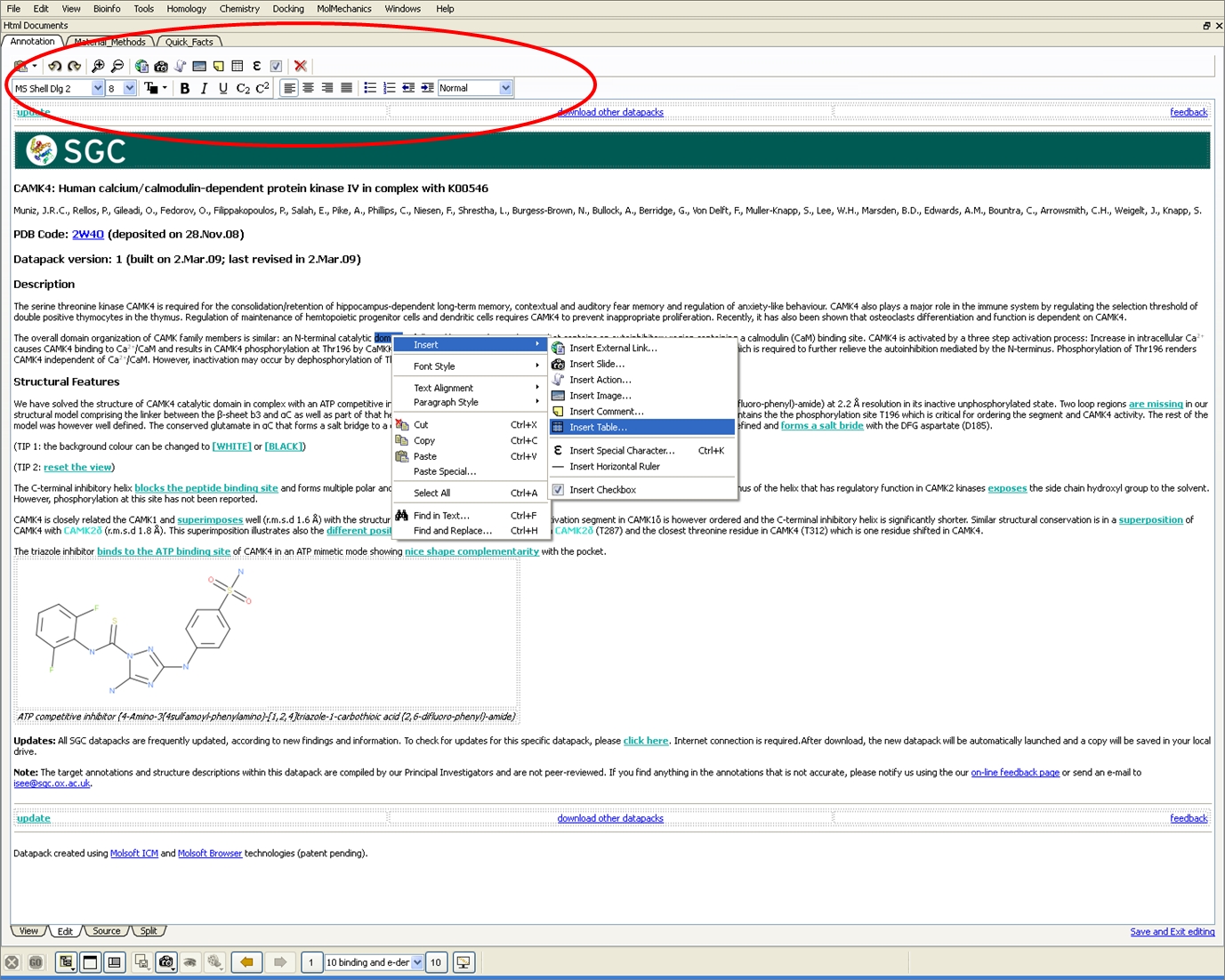
HTML WYSIWYG Editor
The new HTML WYSIWYG editor provides an interface which resembles how the page will be displayed in a web browser - no knowledge of HTML is required!
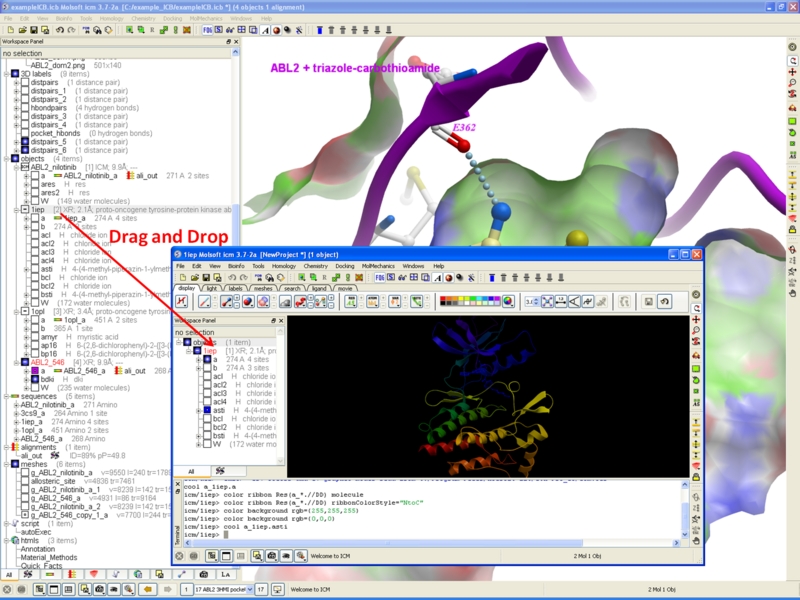
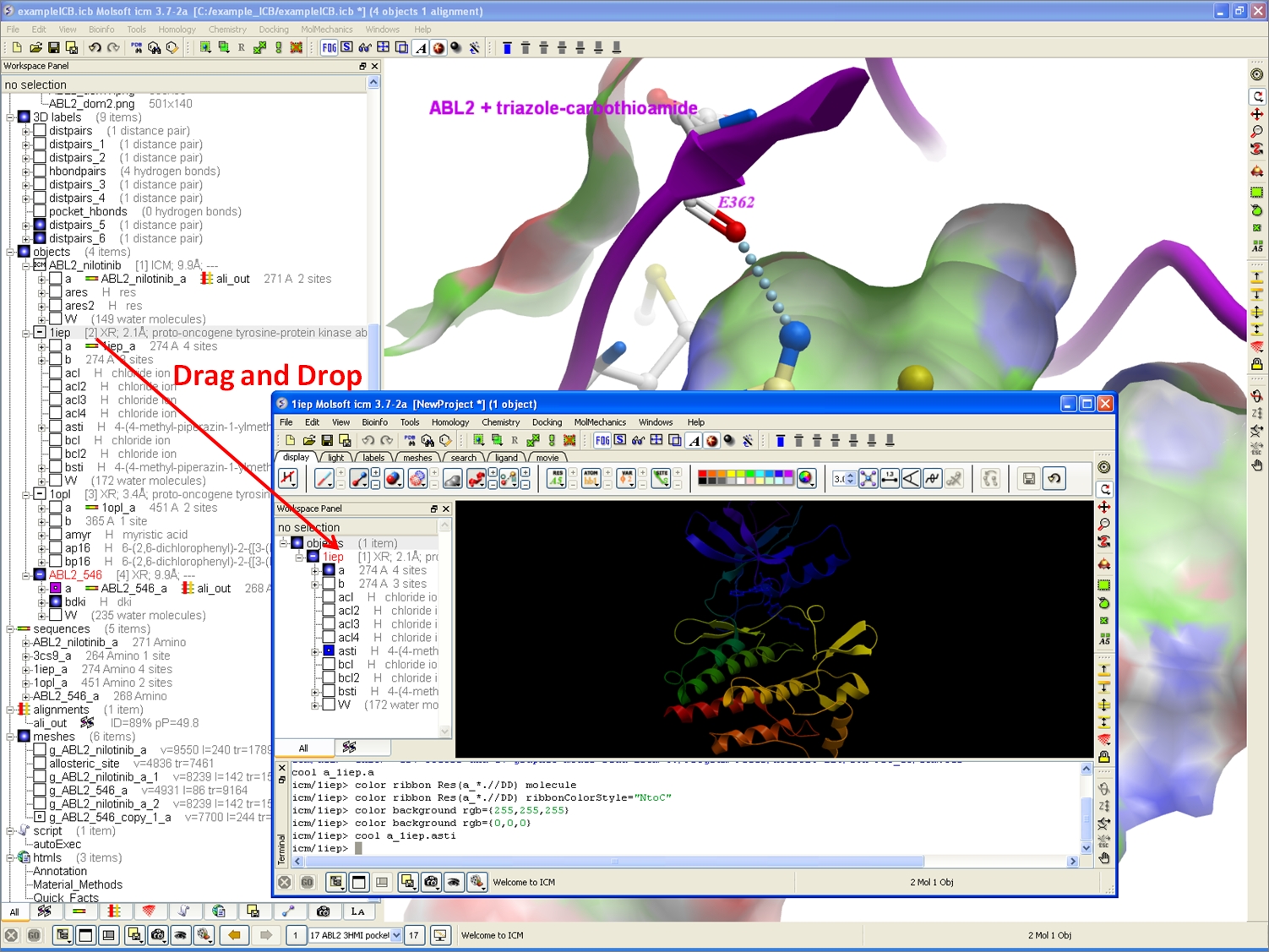
Drag & Drop Copy & Paste
Copy and Paste and Drag and Drop Between Different ICM Sessions.
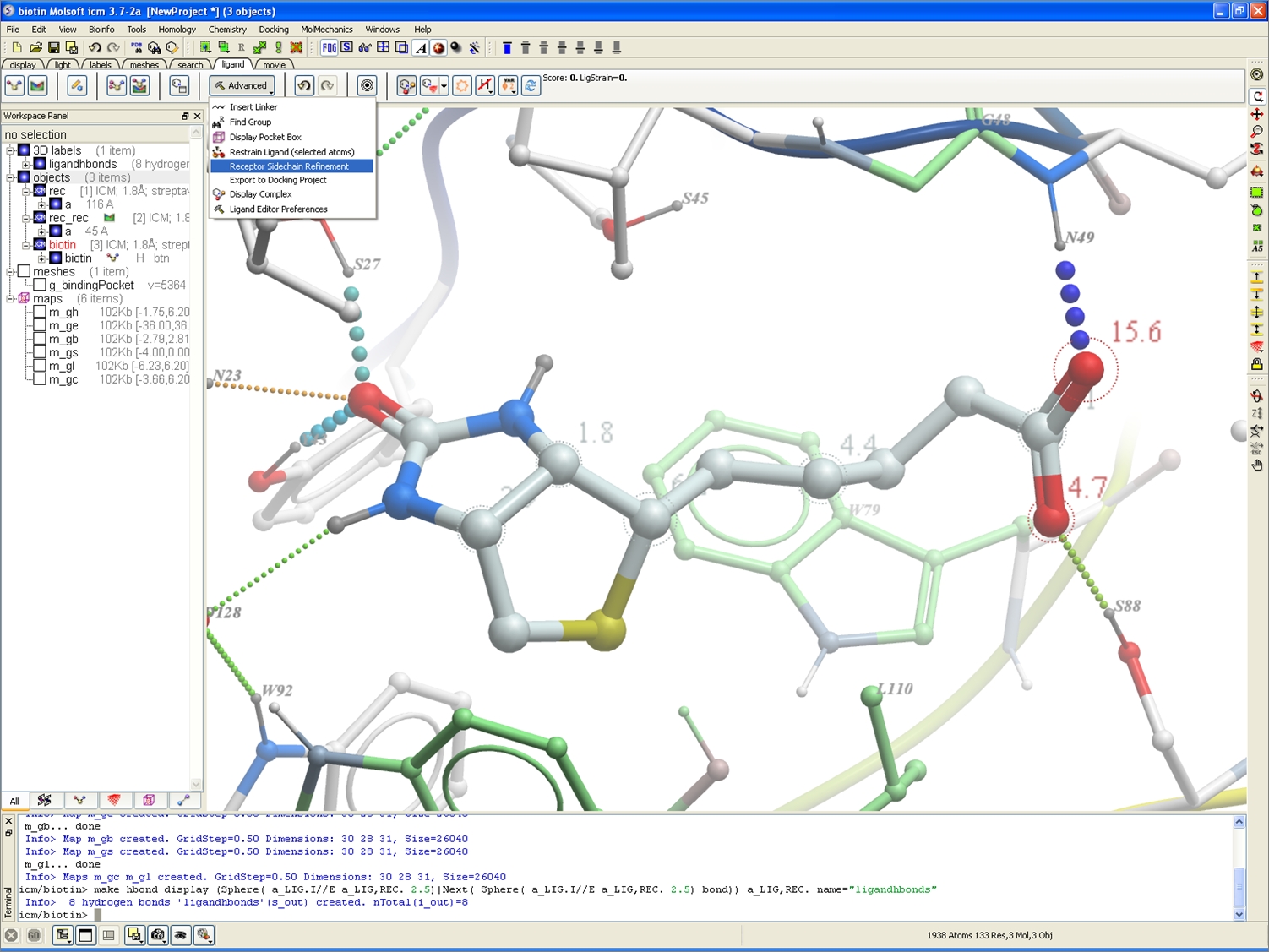
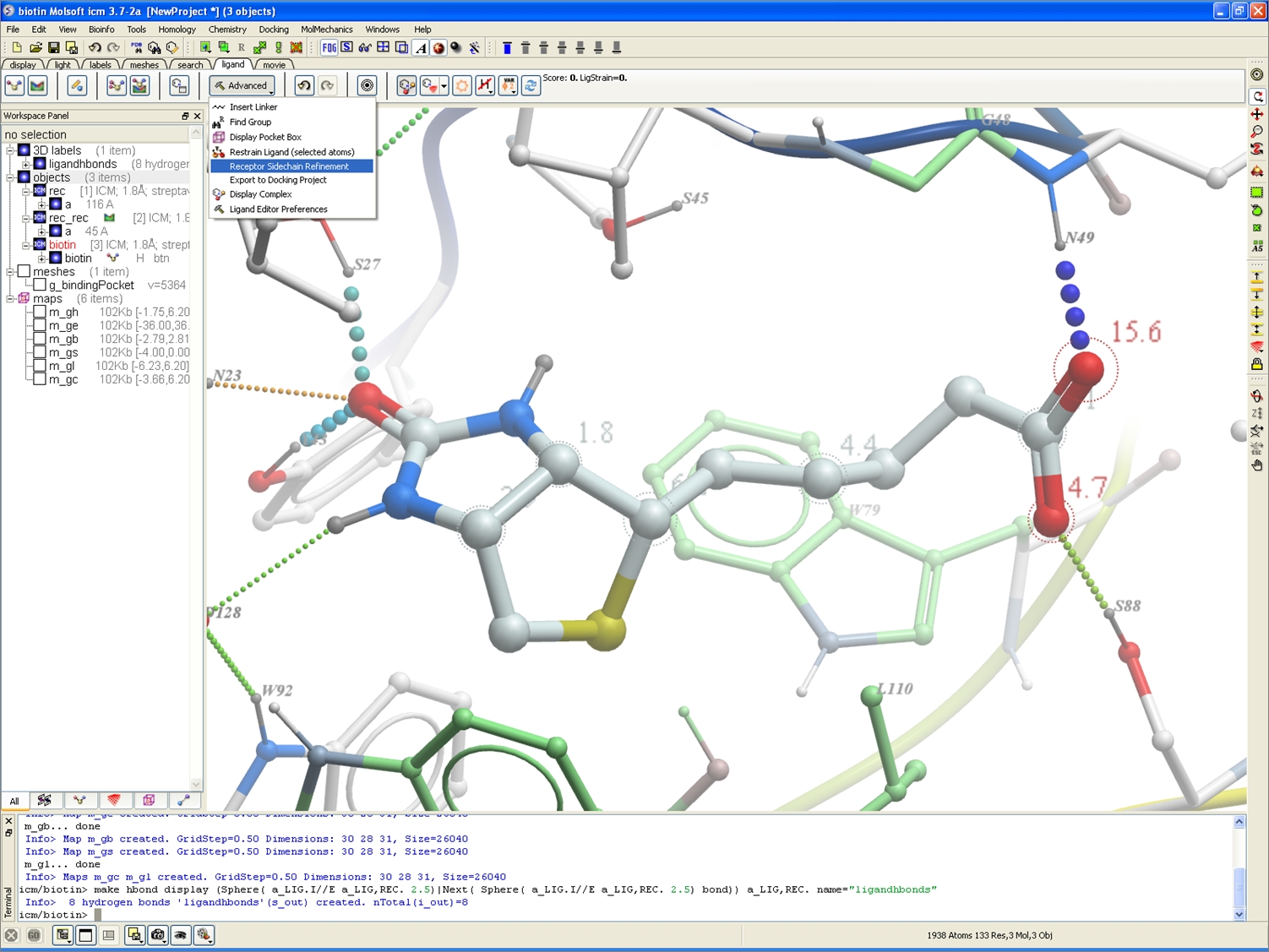
Receptor Refinement
A new option is available in the ligand editor to incorporate receptor side-chain refinement once a chemical modification is made.
CPK Clip Capping
A new preference to control the way CPK spheres are displayed when cut by a clipping plane.
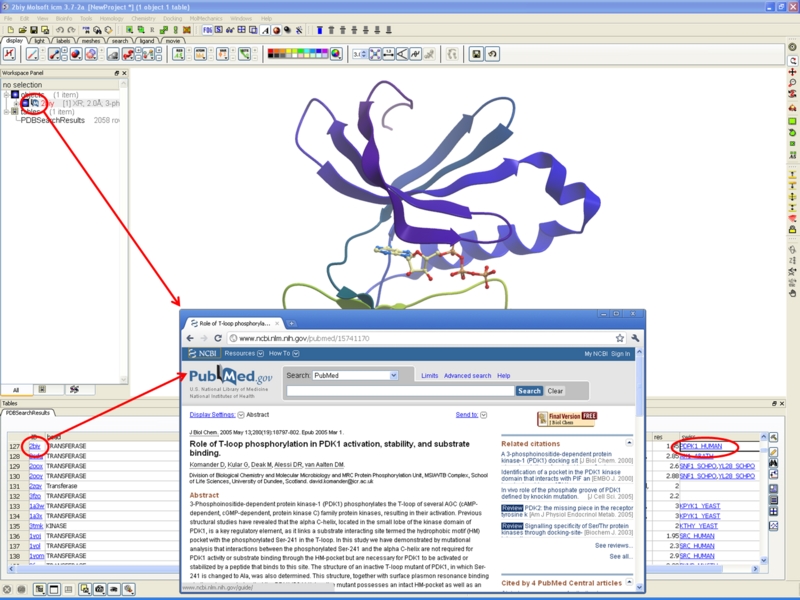
Direct links to PDB and UniProt Websites
New icons and new ways of including external and internal links into ICM tables.
New ICM Force Field
Arnautova et al Proteins 2010
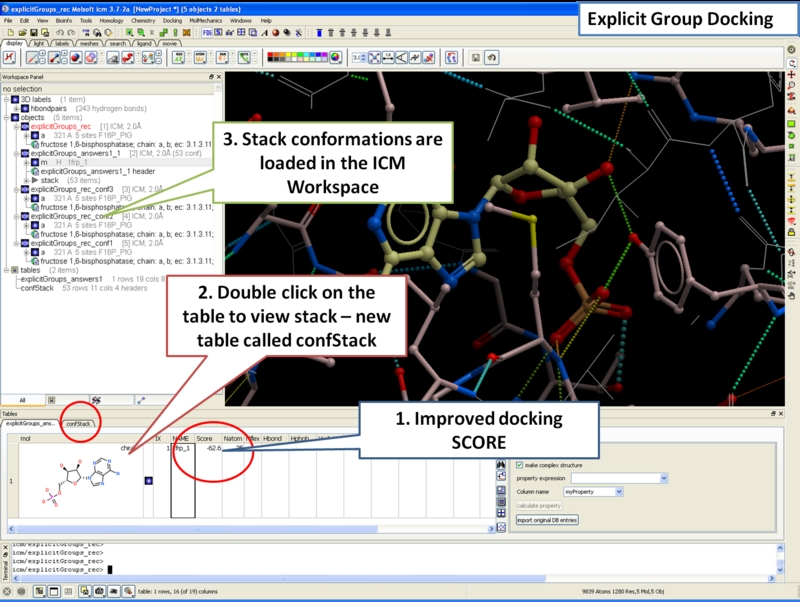
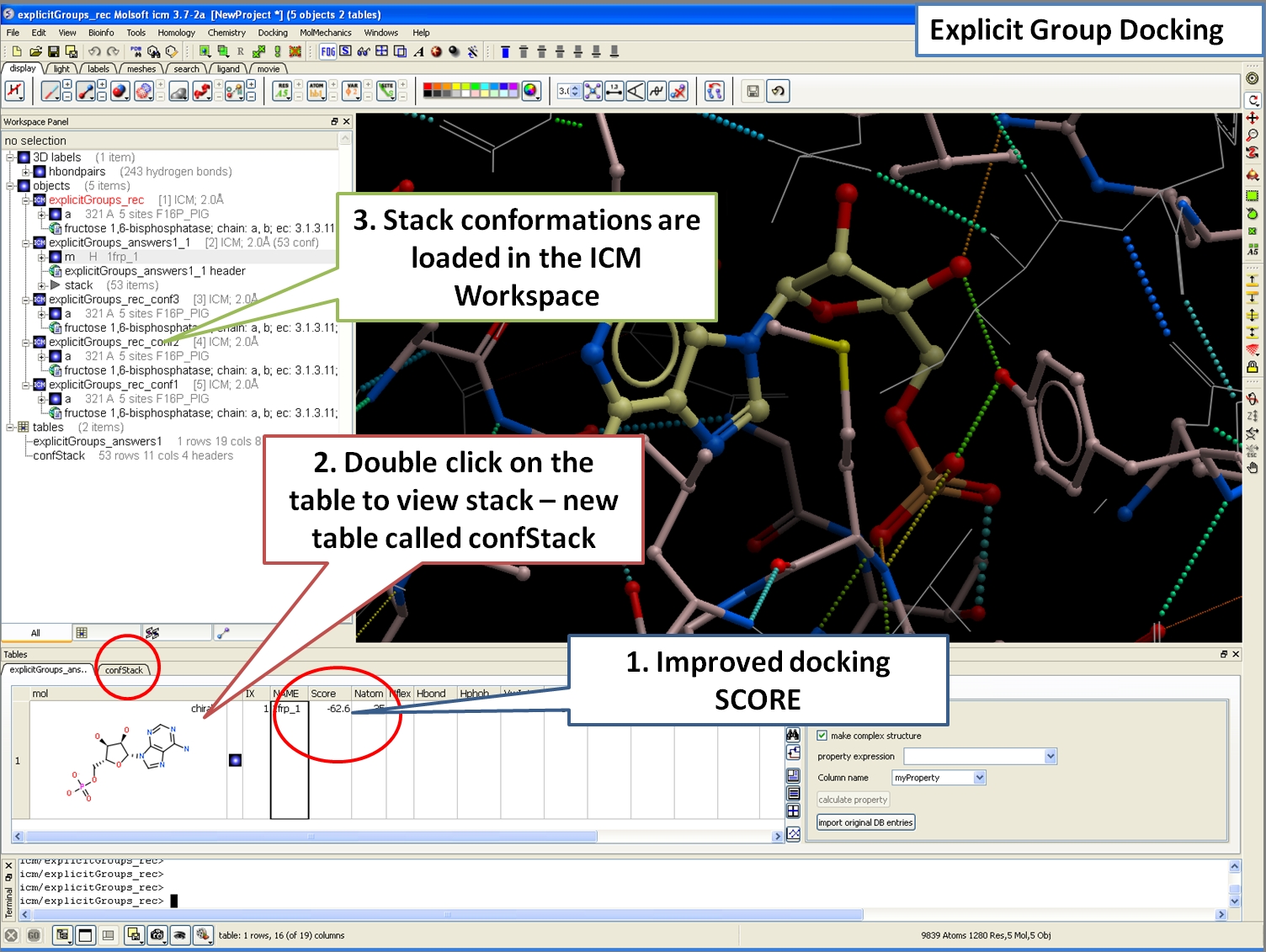
Explicit Group Docking
This new feature allows you to define flexible receptor residues during docking or virtual screening.
New ICM Force Field
The new force-field contains new parametization for the dielectric constant, an improved hydrogen bond determination method, and implementation of novel backbone atom torsional potentials which include bond anlges of the
carbon (alpha) atoms into the internal variable set. Please see the publication in the Proteins journal by Arnautova et al where the force-field has been applied to protein loop modeling.
Follow these instructions to use the new force field in your own scripts for energy minimization and optimization. If you are interested in modeling loops you can use the _loopmodel macro where all these parameters are already pre-set.
Use ICM version 3.7-2a. This can be downloaded by logging into our support site.
The polypeptide must be built or converted with the icmff residue library to do this:
LIBRARY.res = {"icmff"}
readlibrary
The dielectric constant should be set to 2.
dielConst = 2.
In addition to regular energy terms, "bb" must be turned on
setterm"bb"
The force-field method should be set to icmff
ffMethod = 4
New HTML Editor for Molecular Documents
A new HTML WYSIWYG editor which provides an interface which resembles how the page will be displayed in a web browser - no knowledge of HTML is required!
Insert checkboxes with full integration with ICM molecular slides.
Insert ICM scripts.
View HTML source and View HTML output.
WebKit Integration
WebKit has been integrated into the latest ICM version. This will allow:
Full HTML support including (CSS,JavaScript,input elements).
Bridge between ICM scripting language and JavaScript.
Graphics and User Interface
A new preference to control the way CPK spheres are displayed when cut by a clipping plane.
Additional types of occlusion shading have been added (TOOLS.occlusionColorStyle = "dark outside" or "light outside").
Anti-aliased and scalable fonts for atom and residue labels have been added for Windows and Mac.
You can now copy and paste and drag and drop between different ICM sessions.
Option to restore recent backup (see Edit menu).
Tables - a new option called set column format allows support of internal and external links (PDB and UNIPROT).
Translation by the lower screen region.
Docking and Virtual Ligand Screening
Explicit group docking allows you to define specific residues to be flexible upon ligand binding. See an example here.
New option to scan probe fragments.
Chemistry
Charge/protonation state prediction using pKa model.
New Features Added to the Free ICM-Browser
Build and save fully-annotated and interactive 3D molecules and display them in Safari, Firefox, Google Chrome, and Internet Explorer.
Display fully interactive molecules in Windows PowerPoint.
Display ligand pocket surfaces colored by binding property.
Add and optimize hydrogens to a PDB structure.
Calculate and display hydrogen bonds.
Generate and display molecule surfaces.
Display transparent surfaces.
Save publication quality molecular graphics files in a variety of image formats.
3D Interactive Ligand Editor
New option to incorporate receptor side-chain refinement (see the Advanced button in the ligand tab).
New option to export receptor and ligand setup to a docking project (see the Advanced button in the ligand tab). The project is then setup for small molecule docking and virtual ligand screening using the options in the Docking menu.
Implicit Membrane Models
Define shapes with lipids or water for implicit solvation calculaions using TOOLS.membrane array. Each atom is considered as either buried by other explicit atoms of the object or exposed. When the surfaceMethod is defined
as membrane then each atom can be either in water environment or in lipid environment. The way to define the membrane models is discussed in more detail here.
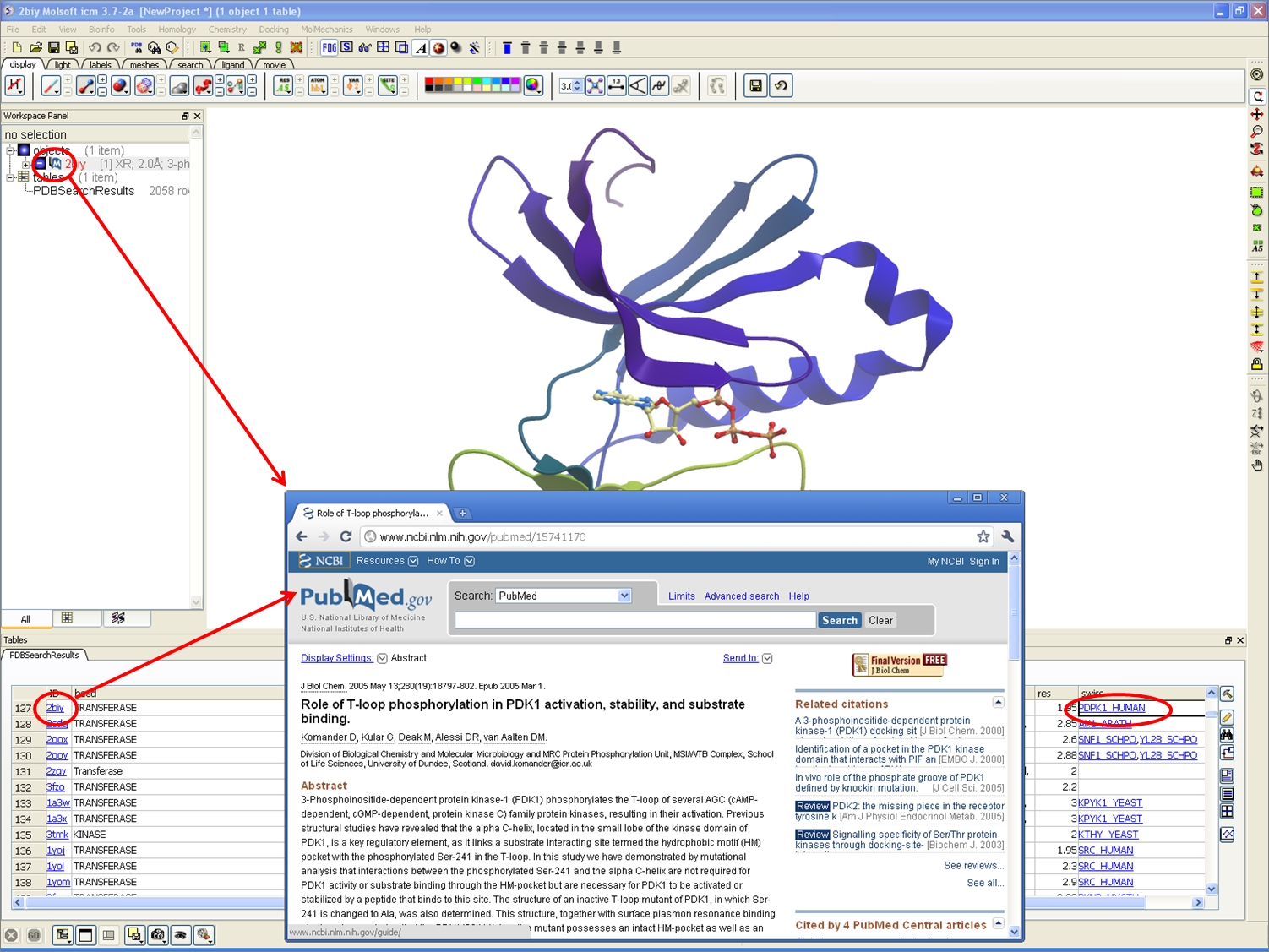
PDB Search
A new icon in the ICM Workspace is now available with a direct link to PubMed icon. Click the icon and you will be taken to the primary reference relating to the displayed structure on the PubMed website.
The PDB Search tab has additional options for displaying NMR models directly from the PDB search. Use the drop down button to determine whether you want to display the first NMR model, all models or all models in a stack.
The PDB Search tab has additional options for displaying occupancy. You can use these options to control l if and how the partial or zero atom occupancies are displayed.
An new option is available when converting a PDB file into an ICM object. You can choose whether or not to sample His, Cys, Pro, Gln, and Asn.
Sequences and Alignments
New sequence annotation style in alignments.
UniProt literature search output.
Tools Menu
Tools/Superimpose by Specific Interaction pair
Tools/Plot/Extras Plot 2D
Tools/Table/Sort Table
Tools/Change Working Directory
MolMechanics Menu
MolMechanics/GAMESS - Link to The General Atomic and Molecular Electronic Structure System (GAMESS) is a general ab initio quantum chemistry package.
MolMechanics/ Generate NM stack -Normal Modes can be used to generate an ensemble of protein structures. For example the method can be used to represent flexibility in the pocket.
MolMechanics/Set Tethers
ICM Shell
Single quote string constant support.
New 2D compound rendering option: color rectangles on hetero atoms instead of atom labels. See set-property-chemical-view
ND support for individual real values
Collection (has table) data structure was introduced.
Additional new features and improvements can be found in our Release Notes.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}