|
|

The Human Cyclophilin Family of Peptidyl-Prolyl Isomerases
Tara L. Davis 1,2,3 , John R. Walker 1 , Valérie Campagna-Slater 1 , Patrick J. Finerty, Jr. 1 , Ragika Paramanathan 1 , Galina Bernstein 1 , Wolfram Tempel 1 , Hui Ouyang 3 , Wen Hwa Lee 4 , Elan Z. Eisenmesser 5 , and Sirano Dhe-Paganon 1,2
1 Structural Genomics Consortium and 2 Department of Physiology, University of Toronto, 101 College Street, Toronto, Ontario M5G 1L7, Canada.
3 Current address: Molecular, Cell & Developmental Biology, University of California Santa Cruz, 1156 High Street, Santa Cruz, CA 95064.
4 Structural Genomics Consortium, University of Oxford, Old Road Campus Research Building, Headington, OX3 7DQ, UK..
5 Department of Biochemistry & Molecular Genetics, University of Colorado Denver, Aurora CO 80045.
Correspondence and requests for materials: sirano.dhepaganon@utoronto .ca" > sirano.dhepaganon@utoronto.ca
Abstract
Peptidyl-prolyl isomerases (EC 5.1.2.8) catalyze the conversion between cis and trans isomers of proline. The cyclophilin family of peptidyl-prolyl isomerases is well known for being the target of the immunosuppressive drug cyclosporin, used to combat organ transplant rejection. There is currently great interest in understanding both substrate specificity for these enzymes and design of isoform-selective ligands. However, the dearth of available data for individual family members inhibits attempts to design drug specificity; additionally, in order to define physiological functions for particular cyclophilins definitive isoform characterization is required. In the current study enzymatic activity was assayed for fifteen of the seventeen human cyclophilin isomerase domains. In addition, cyclosporin binding to all seventeen isomerase domains was probed. In order to rationalize the isoform diversity observed, the high-resolution crystallographic structures of seven cyclophilin domains were determined. These models, combined with six previously solved cyclophilin isoforms, provide the basis for a family-wide comparison of structure and correlation with function. Detailed structural analysis of the human cyclophilin isomerase explains why cyclophilin activity against short peptides is absolutely correlated with ability to ligate cyclosporin and why certain isoforms are not competent for either activity. In addition, true isoform specificity for both in vivo substrates and for potential drug design must be encoded by regions of the isomerase domain outside the proline-binding surface. We hypothesize that there is a well-defined molecular surface, corresponding to the substrate binding site at the -2 position, that is a site of isoform diversity in the cyclophilin family. Computational simulations docking substrates into this region provides data that is correlated with experimental data. Our data indicates that there are unique isoform determinants that may be exploited for development of selective ligands and suggests that the currently available small molecule and peptide-based ligands for this class of enzyme are insufficient to encode true isoform specificity.
Introduction
Cyclophilins are peptidyl-prolyl isomerases (PPIases: EC 5.2.1.8) and are characterized by their ability to catalyze the interconversion of cis and trans isomers of proline. Cyclophilins, much like the structurally unrelated FK506 binding proteins, were initially described as the in vivo receptors for the natural products cyclosporin, FK506/tacrolimus, and rapamycin/sirolimus [1-3]. The immunosuppressant effect of these natural products, while revolutionizing the field of organ transplantation, were eventually determined to be unrelated to the inherent isomerase activity of the PPIases [4]. However, these small molecules bind to the active site of PPIases with high affinity and are capable of blocking isomerase activity against peptide substrates, making them a useful tool for biochemical and cellular assays of PPIase function [5].
For many years it has been theorized that the physiological function of the PPIase activity of the cyclophilins is as a chaperone or foldase [2,6]. Certainly this functionality is well documented, for instance in the maturation of steroid receptor complexes (along with Hsp90/Hsc70) [7] or in the interplay between NinaA and rhodopsin in Drosophila [8]. In addition, the isomerase activity of at least two cyclophilin isoforms is crucial for host:viral interactions and for viral maturation processes, and this activity seems to be mediated through the PPIase active site [9,10]. However, it has become apparent that PPIases also function as signal transducers, and that this function is separate from a foldase or chaperone activity. The most obvious example in the literature of this is the non-immunophilin Pin1, a PPIase of the parvulin type. Pin1 is only able to catalyze isomerization of the proline bond for target substrates when a serine or threonine preceding the target proline is phosphorylated [11]. This phosphorylation-dependent isomerization has wide-ranging effects on the cellular signal pathways that Pin1 is involved in, including proliferation and tumorigenesis [12]. The identification of Pin1 substrates revitalized the search for additional functions of the immunophilin-type PPIases; although there is no example of phosphorylation-dependent isomerization for either FK506 binding proteins or for cyclophilins, a subset of substrates for these types of PPIases are certainly also dependent on non-chaperone functions. PPIA, along with classical functions in the chaperone-mediated processes outlined above, interacts with the non-receptor tyrosine kinase Itk and affects its activity state in vivo [13]. PPIA also is known to modulate HIV infectivity by interacting with a proline-containing sequence in the capsid protein Gag [14]. More recently, PPIA has been shown to interact with CD147 in a manner that is proline-dependent and mediated through the active site of the isomerase [15, 16]. In addition, both PPIA and the highly similar PPIB have been shown to interact with NS5B, an RNA-dependent RNA polymerase necessary for hepatitis C viral replication [9, 17]. The three other single-domain PPIases - which encode only the PPI domain, and in the case of PPIB and PPIC a signal sequence - and the thirteen multidomain PPIases are less well-characterized; most of what is known for these cyclophilins is uncoupled from their isomerase activity. For instance, the single domain PPIase PPIH (SnuCyp20) participates in the spliceosome through interactions with the 60K component of the tri-snRNP, also known as PRP4; however, the co-crystal structure of PPIH with a peptide derived from PRP4 showed that this interaction was mediated exclusively through a face opposite that of the active site [18]. A similar situation was found in another single domain spliceosomal cyclophilin, PPIL1, which interacts with the protein SKIP; NMR data indicates that the chemical shift perturbations in PPIL1 upon SKIP binding did not involve residues involved in prolyl turnover [19]. Finally, PPIE has an RNA-recognition motif (RRM) and has been reported to have RNA-specific isomerase activity [20].
Cyclophilins have been implicated in diverse signaling pathways, from the relationship of the mitochondrial cyclophilin PPID to the permeability transition pore [21, 22], to the spliceosomal cyclophilins [23,24], to the transmembrane NKTR and its involvement in NK triggering [25]. However, these studies have not identified the particular proteins involved. Specific non-chaperone type activities for the cyclophilins are difficult to find, at least in part because very little about the biochemistry of these proteins have been described. For instance, of the seventeen human cyclophilins only seven have been tested for isomerase activity or for the ability to bind cyclosporin [19, 26-31]. In vitro techniques aimed at delineating substrate specificity for the canonical family member PPIA have only been moderately successful; in the case of phage display, the optimized binding sequence does not correspond to the substrate determinants that have been found in vivo for this isoform, and this sort of randomized screening has not been accomplished for any of the less ubiquitous isoforms [32]. Generally, the issue of in vitro versus in vivo substrate selectivity for the isomerases is a vicious circle: since the isomerase substrate specificity in vitro is not delineated for all isoforms, it is difficult to find and validate in vivo substrates for these enzymes; and for the isoforms that have been tested in vitro, there is little predictive value in finding in vivo substrates. Clues in some cases may be derived from the identity of other domains expressed in tandem with the cyclophilin domain; for instance, the RRM domain previously mentioned implies an RNA targeting function for PPIE and PPIL4, and likewise the U-box motif of PPIL2 implies involvement in ubiquitin conjugation pathways [33]. The WD-40 repeat of PPWD1 most likely confers a protein:protein interaction function, as this is its main function in other systems; the same holds true for the TPR motifs of RanBP2 and PPID. However, true comparisons of in vitro activity with in vivo physiology must wait until the cyclophilin family is more fully characterized along both lines of research.
In this study, we have determined the structures of seven PPI domains to high resolution using X-ray crystallography and characterized the human cyclophilin family with regards to their activity against standard substrates and ability to bind the natural product. These studies show interesting structural and enzymatic diversity among the members of the cyclophilin family that is well explained by the structural data. The structures also provide an opportunity to assess the cyclophilin family for regions of diversity among all family members, and in silico methods were used to characterize a molecular feature contiguous with the canonical active site that may account for substrate specificity.
(TIP 1: the background colour can be changed to [WHITE] or [BLACK] )
(TIP 2: reset the view )
Results/Discussion
Characterization of Cyclophilin Active Sites: In order to elucidate the function of residues in the extended active site of the PPI domain of the human cyclophilins, we probed the binding and catalytic function of the PPI domains against substrate or small molecule inhibitors. Two assays were utilized to explore these functions: in the first assay, changes in thermal stability were used to assess cyclosporin binding. This assay has previously been shown in several studies to be a reliable readout of small molecule binding for kinase and enzyme families [34-36]. Cyclosporin A and the derivatives cyclosporin C, D, and H were screened against all PPI domains except for PPIL4, for which all constructs were insoluble in our hands (Table 1). Due to the inherent thermal stability characteristics of PPID, PPIL3, and RanBP2, this technique was unable to distinguish between apo and cyclosporin bound forms of those domains. However, acceptable data was collected for all the remaining thirteen isoforms, and binding to at least one of the cyclosporin derivatives was noted for six isoforms published previously including PPIA, PPIB, PPIC, PPIE, PPIL1, and PPWD1 [19,26-29,31]. In addition, binding of cyclosporin A or derivatives was seen for PPIF, PPIG, PPIH, and NKTR. In the case of PPIG and PPIH, this explains previous data describing cyclosporin binding to the tri-snRNP complex that contains PPIH [24] and verifies the finding from a homolog that PPIG is capable of binding cyclosporin [37]. To our knowledge, this is the first set of experiments showing cyclosporin binding to any homolog of PPIF, PPIL1, or NKTR. Surprisingly, no binding was detected for PPIL2, PPIL6, or SDCCAG-10 (Table 1).
A two-dimensional NMR experiment (1H/1H TOCSY) described previously [29,38] was used to assess activity on commercially available tetrapeptides AAPF, AFPF, and AGPF (all modified with an N-terminal succinyl and C-terminal p-nitroanilide moiety). These substrates are generally used in the field for in vitro characterization of isomerase activity [39]. The NMR experiment is a sensitive method to probe for both substrate binding and catalytic activity where it exists. In addition, it is not dependent upon protease-coupled protocols and is straightforward to reproduce with unlabeled protein and/or peptides. In our hands, there is clearly binding and turnover for at least one of the tetrapeptide substrates tested for PPIA, PPIB, PPIC, PPID, PPIE, PPIF, PPIG, PPIH, PPIL1, PPWD1, and NKTR (Table 1; see Figure S1 for examples of positive and negative activity). This correlates well with previously determined activities [1,19,20,27,29,40,41], and also establishes activity measurements for PPIF, PPIG, and NKTR. For all isoforms tested there was a strict correlation between the ability to bind cyclosporin and activity against the tetrapeptide substrates (Table 1).
In order to understand the molecular basis of these results, structural coverage of the entire human cyclophilin enzymatic class was sought. We determined crystal structures of seven human PPI domains, which together with six previously determined structures leave only two uncharacterized human cyclophilins. The thirteen experimentally determined structures - plus the highly homologous bovine PPID (three amino acid substitutions compared to human) - globally very similar secondary structural elements, and therefore provide excellent homology models for the remaining three isoforms (PPIL4, PPIL6, and the PPI domain of RanBP2). Models for these three isoforms were generated using the Phyre algorithm [42], and for all further discussions of the cyclophilin family the structures of all seventeen PPI domains will be considered.
All cyclophilins share a common fold architecture consisting of eight anti-parallel β sheets and two α-helices that pack against the sheets (Figure 1). In addition there is a short α-helical turn, containing the active site residue Trp121 (all residue identities and residue numbers correspond to PPIA except where noted), found in the β6-β7 loop region (Figure 2). An overlay of the Phyre-derived modeled structures leads to an RMSD over all atoms of 1Å or less compared to PPIA. The PPI fold can be considered to be conserved as RMSD across all atoms for all PPI domains are less than 2Å, and sequence identity over the same region varies from 86% to 61% (Figure 1 and Figures S2 and S3). The most divergent structures in this set are PPIL1, which is an NMR-derived structure (RMSD 1.7Å), and the previously described PPWD1 (RMSD 1.4Å) [29]. The rest of the PPI domains align with RMSD over all atoms ranging from 0.4 Å - 1.0 Å (Figure 1 and Figure 3).
|
|
|
Figure 1. Structural elements of the cyclophilin fold and the definition of the active surface of PPIA. (A) Secondary structural elements of PPIA in ribbon representation, with key structural elements labeled. All structural outputs were generated using PyMol unless otherwise noted. (B) Consurf representation of sequence conservation within the human cyclophilin family; residues that comprise the active surface of the cyclophilin family are labeled [73]. (C) The molecular surface of the PPIA structure. The view is oriented roughly as in (A) and (B). (D) Comparison of the sequences that define the active surface of the PPI domain. Residue numbering corresponds to PPIA. |
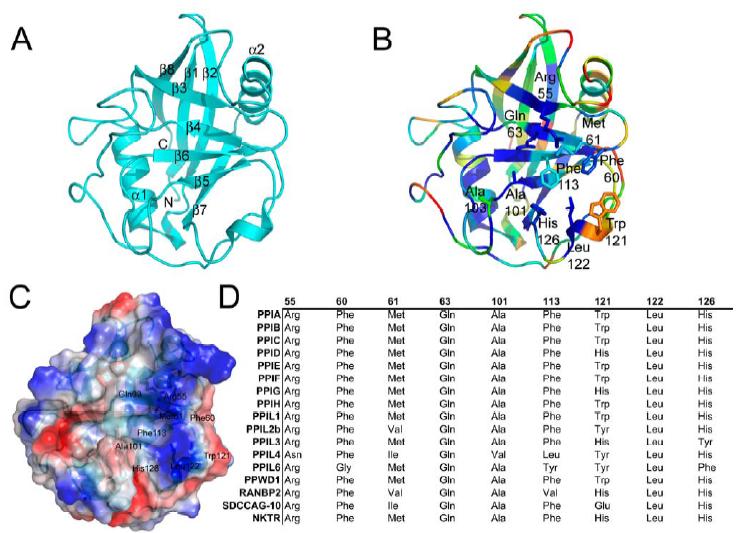
The active site of the cyclophilin family includes the invariant catalytic arginine ( Arg55 ) and a highly conserved mixture of hydrophobic, aromatic, and polar residues including Phe60 , Met61 , Gln63 , Ala101 , Phe113 , Trp121 , Leu122 , and His126 [43-45]. All of these side chains contribute to an extensive binding surface along one face of the PPI domain measuring roughly 10 Å along the Arg55-His126 axis and 15 Å along the Trp121-Ala101 axis (Figure 2). Many of these residues are well conserved across all PPI domains and are thought to serve functions in either catalysis or substrate/inhibitor binding [43,45,46] (Figure S2). Although there are sites of minor diversity among the family members at the Phe60, Met61, and His126 positions, the most striking correlation between cyclosporin binding, tetrapeptide identity, and active site residues is found at the Trp121 position. Our results clearly show that a tryptophan (as found in PPIA , PPIB , PPIC , PPIE , PPIF , PPIH , PPIL1 , and PPWD1 ) or histidine (as found in PPID , PPIG , PPIL3 , RANBP2, and NKTR ) at this position is permissive for cyclosporin binding whilst other naturally occurring residues at this position (tyrosine in PPIL2, PPIL4, and PPIL6, and glutamic acid in SDCCAG10) abrogate cyclosporin binding (Table 1 and Figure 2). It is well known that mutating Trp121 in PPIA to alanine or phenylalanine has a negative impact on cyclosporin affinity [46-48]. Mutation of the naturally occurring histidine in PPID to a tryptophan increases cyclosporin affinity 20-fold, although the wild-type enzyme was capable of binding cyclosporin with low micromolar affinity [30, 49]. More current work attempting to elucidate the mechanism of action of PPIA has utilized computational modeling trials, with the results indicating that the function of Trp121 is mainly to serve to build a hydrophobic pocket for the substrate proline to interact with (along with Phe60 , Met61 , Phe113 , and Leu126 ) [50,51]. There are no mutational or computational data for the cyclophilins that have a tyrosine or glutamic acid substitution at the Trp121 position; but at least in the case of PPIL2, which is identical to PPIA at every active site position save a conservative Met61-Val substitution, it seems clear that the lack of cyclosporin binding is associated with the tyrosine substitution at the Trp121 position.
|
|
|
Figure 2. The structural consequences of substitutions in the cyclophilin active site. The residues described in Figure 1 are shown in stick representation for the divergent family members PPIA, NKTR, PPIL2, and SDCCAG-10. Note the orientation of the divergent residues Tyr389 in PPIL2 and Glu122 in SDCCAG-10 relative to Trp121 in PPIA or His132 in NKTR. |
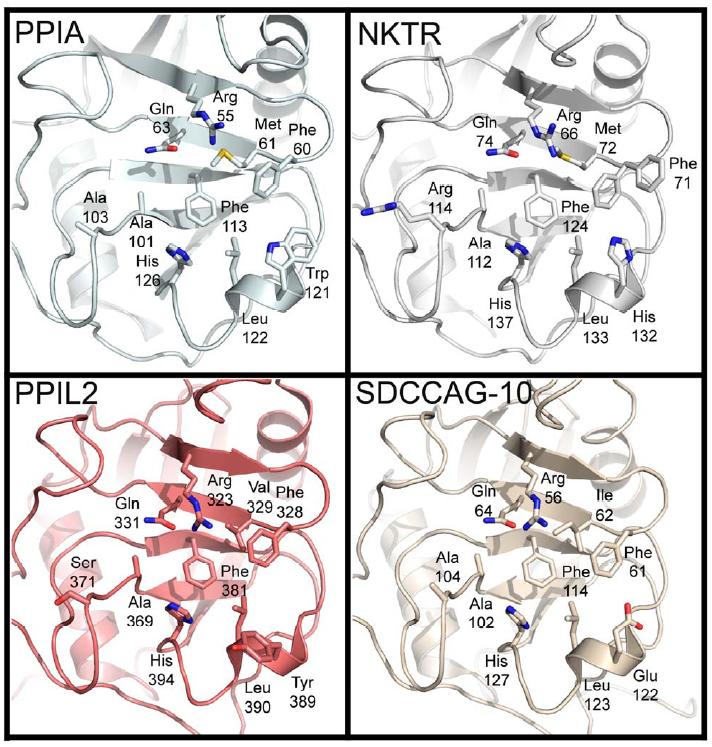
Modeling of either CsA into the active site of a histidine containing isoform (like NKTR) or computational mutation of the Trp121 in a PPIA:CsA complex structure indicates that similar hydrogen bond distances can exist between the indole moiety of tryptophan or the imidazole ring of histidine and the carbonyl of methylleucine 9 (MLE9) in CsA (Figure 2 and Figure S4). Therefore either residue would be competent for binding, as we have shown experimentally. Conversely, modeling a tyrosine into the conformation necessary to interact with CsA shows clearly that this would create a steric clash with MLE9; in addition, there is a close steric conflict with the modeled Tyr residue and the highly conserved Phe60 residue that helps form the proline binding pocket (Figure S4). Perhaps this is why in our apo PPIL2 structure the tyrosine at this position points away from the active surface (Figure 2). Taken together, our results indicate that the indole ring of tryptophan or the imidazole sidechain of histidine are the most important determinants of cyclosporin binding, and cannot be substituted for by other sidechain chemistries.
There are three cases in which we can detect neither cyclopsorin binding nor tetrapeptide binding: PPIL2 , PPIL6 , and SDCCAG-10 (Table 1). It is clear that these three proteins are quite divergent in their active surface compared to PPIA (Figure 1D). Perhaps more importantly they are, along with PPIL4, the only isoforms that substitute the residue Trp121 with a non-histidine residue. Additionally, PPIL6 does not encode the otherwise strictly conserved Arg55 (there is an asparagine at the equivalent position), so it is not surprising that this isoform does not show activity against standard substrates. There is no literature concerning the function of the PPI domain for these inactive isoforms, but it is interesting to note that that structurally these isoforms could still serve as a proline-binding motif; or alternatively these PPI domains are specific for substrates not represented by our test set.
|
|
|
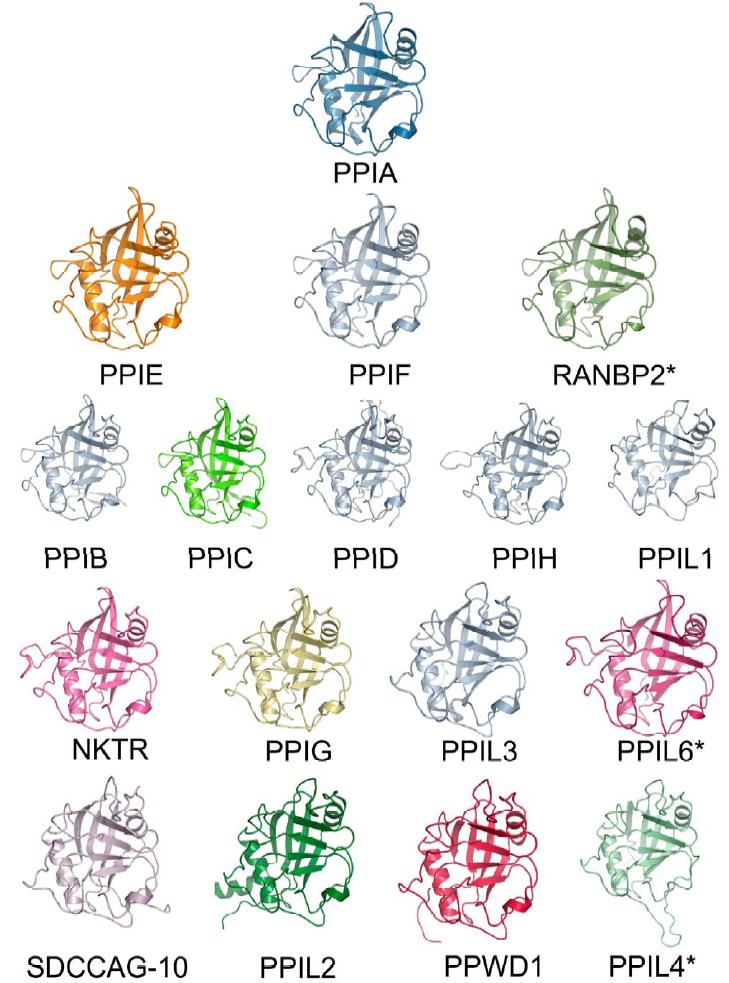
Figure 3. Structural coverage of the human cyclophilin family. Cartoon representation of the experimental and modeled structures of human cyclophilins. Only the isomerase domain is shown. Structures solved previously are shown in shades of grey, while the novel structures associated with this manuscript are colored. The structures of RanBP2, PPIL6, and PPIL4 are marked with an asterisk, as they are derived from homology modeling using the Phyre server [42] and do not represent experimentally derived data. The structures are arranged roughly according to their similarity to PPIA by sequence alignment (See Figure S3). |
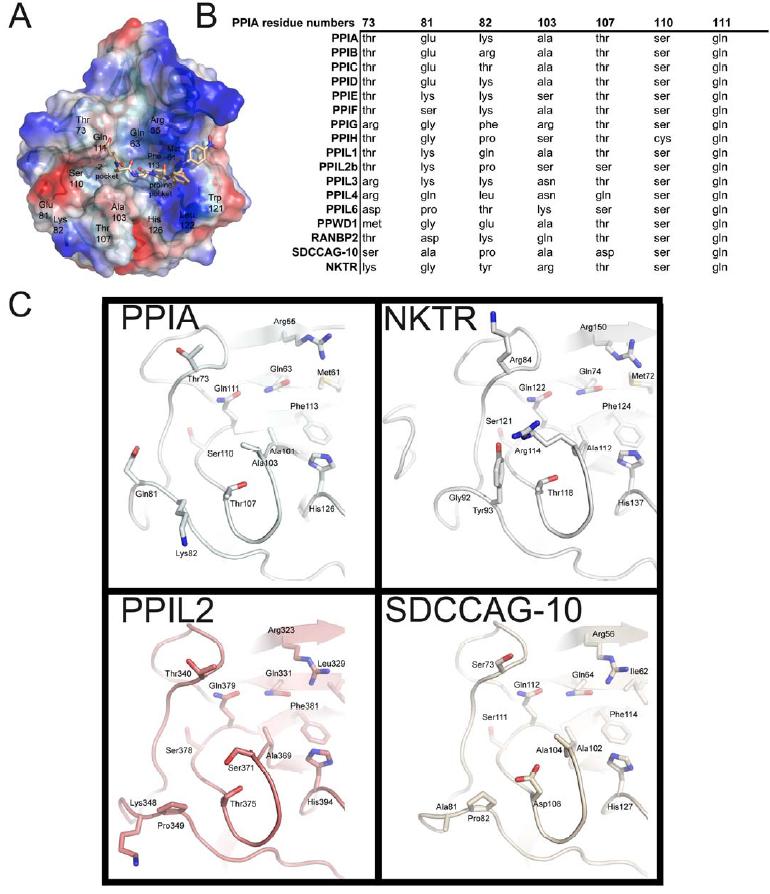
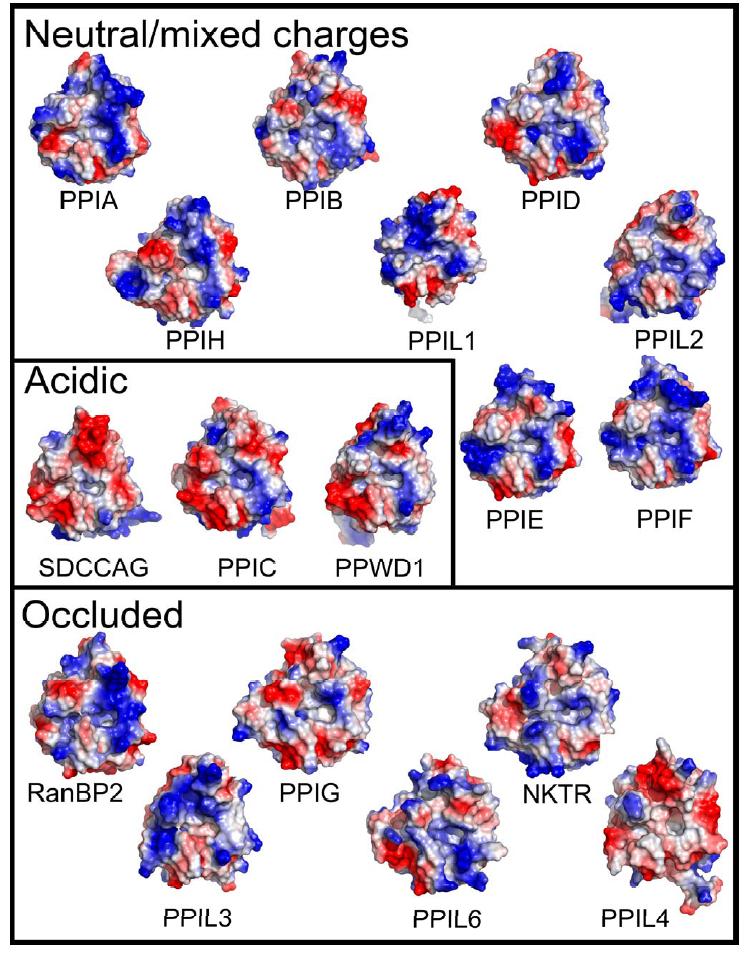
Although the cyclophilin fold is highly conserved, we next sought to find potential sites of substrate selectivity by undertaking a more extensive structural analysis of the cyclophilin family. We found by examining the surface of the PPI domains near the active site that there are two pockets that potentially contribute to substrate specificity, binding, and turnover. The first pocket , which is the proline interaction surface, is defined by the PPIA residues Phe113 which forms the base of the pocket; and Phe60 , Met61 , Leu122 , and His126 which form the sides of the pocket (Figure 1 and 2). As previously discussed, these residues are highly conserved across most PPI isoforms and most species that encode cyclophilins (Figure S2) [52]. The second pocket forms a surface that should interact with substrate residue -2 or -3 relative to the substrate proline, and so will be named the -2 pocket hereafter. Unlike the proline pocket, it is the main-chain atoms of the β5-β6 loop that define the base of the -2 pocket. As a consequence, the chemical identities of residues found in this region do not have much influence on the size and shape of the -2 pocket (Figure 4). Indeed, the -2 pocket is extremely uniform across the cyclophilins; it seems to be deep and relatively nonspecific, so that it can accommodate long, short, polar, or hydrophobic sidechains without penalty. However, the -2 pocket surface is guarded by a set of "gatekeeper" residues whose sidechains are in a position to control access to this pocket. In PPIA , these residues are Thr73 , Glu81 , Lys82 , Ala103 , Thr107 , Ser110 , and Gln111 (Figure 4). These gatekeeper residues at positions 81, 82, and 103 and the secondary gatekeeper at position 73 (called a secondary gatekeeper because its position in most PPIase structures is pointed away from the -2 pocket) show major chemical and size variance. For instance, the residue that is at position 103 in PPIA varies from alanine in about half of the cyclophilin isoforms and a serine in PPIE , PPIH , and PPIL2 ; to an arginine in PPIG and NKTR , lysine in PPIL6 , asparagine in PPIL3 and PPIL4 , and glutamine in RANBP2 (Figure 4). The identities of the amino acids at positions 73, 81 and 82 are equally diverse across the cyclophilin family. The practical effect of this variance can be visualized by examining the surface properties of the cyclophilin family (Figure 5). These surfaces are clearly unique to the individual cyclophilin members, but can generally be classified into gatekeeper surfaces with mixed or neutral charges (for example, PPIA and several others); gatekeeper surfaces with overall acidic character (SDCCAG-10, PPIC, and PPWD1); and gatekeeper surfaces that occlude access to the -2 pocket (several; see Figure 5). The occluded set of surfaces includes many of the cyclophilin isoforms with bulky sidechains at the gatekeeper positions; for instance, the gatekeeper residues for NKTR include Lys84 , Tyr93 , and Arg114 compared to PPIA residues Thr73 , Lys82 , and Ala103 (Figures 4 and 5). Finally, residues within this region of PPIA have previously been shown to exhibit a functional dependence on binding using NMR relaxation studies [53], consistent with our gatekeeper hypothesis. Thus, the functional significance of these unique -2 pocket surfaces will be further explored in regard to their effect on substrate binding and specificity.
|
|
|
Figure 4. The -2 “gatekeeper” region of the human cyclophilins. (A) The definition of the proline pocket and -2 pocket is shown by depiction of a complex between PPIA and the tetrapeptide suc-AGPF-pNA (PDB 1ZKF). (B) Comparison of the sequences that define the -2 pocket of the PPI domain. Residue numbering corresponds to PPIA. (C) Sequence diversity of the gatekeeper residues in four PPI domain structures. As shown in Figure 5, these substitutions lead to diverse size and charge properties in this region of the cyclophilin active surface. |
Note that other than around the -2 pocket, there is little conformational deviation throughout the cyclophilin family (Figure 3); most of the remaining structural diversity is found in the loop regions connecting α and β secondary structure. First, there is a set of isoforms that encode a deletion in the β1-β2 loop region (residues Ala11-Pro16 in PPIA) that significantly alters the β sheet lengths in this region along with the loop between them. The division between "deleted" β1-β2 loops and "full-length" β1-β2 loops follows a phylogram distribution of PPI domains, with the more conserved isoforms relative to PPIA ( PPIB , PPIC , PPID , PPIE , PPIF , PPIG , PPIH , PPIL6, NKTR , and RanBP2) encoding full-length loops and the more divergent members by sequence (PPIL1, PPIL2, PPIL3, PPIL4, SDCCAG-10, and PPWD1) encoding deleted β1-β2 loops (Figure 3 and Figures S2 and S5). The area encompassed by the α1-β3 loop ( Thr41 - Gly50 ) is similar in that a subset of isoforms encodes longer and shorter versions of this loop. There are three distinct classes of conformations adopted by the α1-β3 loop in the cyclophilin family: the PPIA α1-β3 loop family , the PPIA α1-β3 loop family, which includes PPIA , PPIB , PPIC , PPIE , and PPIF ; a shorter version of the loop represented by the structures of PPIL1 , PPIL2 , PPIL3 , PPIL4, SDCCAG-10 , and PPWD1 ( PPIA is included for reference); and a longer version encoded by PPID , PPIG , PPIH , PPIL6, and NKTR . This α1-β3 loop diversity changes the orientation of the α1 helix and the β3 sheet, and causes up to a 4-6Å movement of α1 relative to PPIA (Figure S5). Finally, the α2-β8 loop ( Gly146-Lys155 ) encodes two distinct groupings of structures : the standard conformation found in PPIA , PPIE , PPIF , PPIL6, and RANBP2, and the conformation adopted by all other isoforms. From sequence alignment it appears that PPID should adopt the standard conformation, as it aligns more closely with PPIA in that region; however, the structure shows that PPID is more closely related to the non-standard conformation in the α2-β8 loop region (Figure S5). It is interesting to note that two regions found to have structural divergence (the β1-β2 and α2-β8 loops) actually form a contiguous surface on the "back" face of the cyclophilin fold relative to the active site. It is possible that sequence and structural diversity in this region could indicate a preference for different potential binding partners, as the back face of the cyclophilins has been seen previously to modulate protein:protein interactions [18,19]. However, it seems that for substrate interactions mediated by the proline binding pocket, isoform selectivity is likely to be determined by the -2 pocket region rather than the β1-β2/α2-β8 regions.
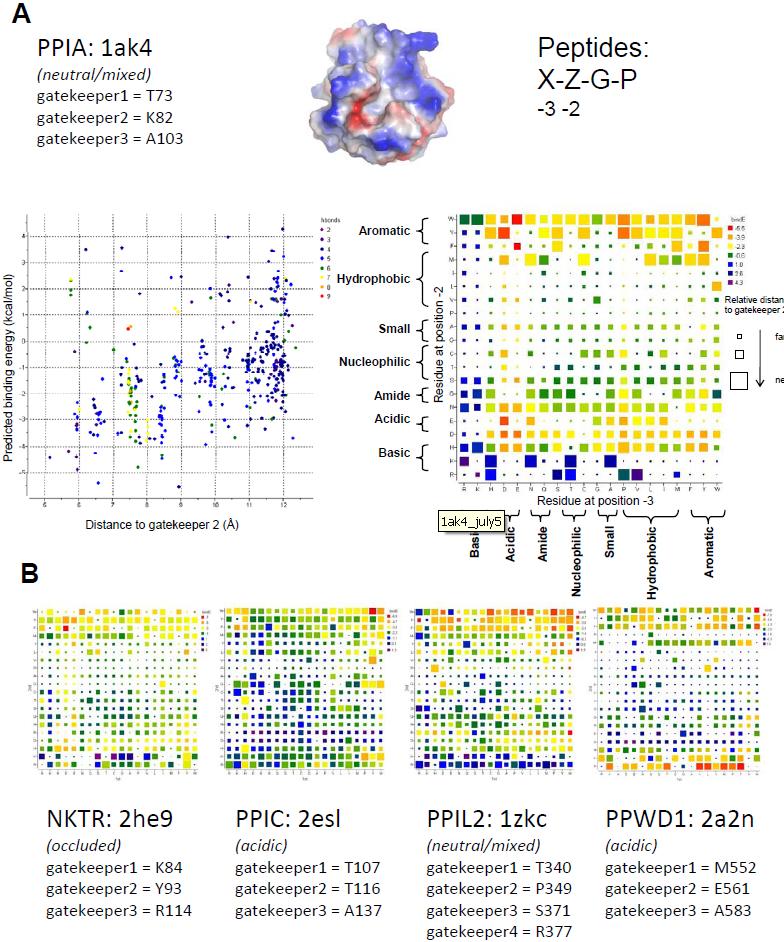
Cyclophilin Diversity in the "-2 Gatekeeper Region": Our biochemical data are the latest evidence that the molecular determinants for tetrapeptide substrate or cyclosporin binding may not be identical to the molecular determinants for physiologically relevant substrates, and supplements other recent publications along these lines [54,55]. Additionally, our structural analysis has led us to the conclusion that the region surrounding the -2 pocket is the most likely spot to target in order to obtain isoform specificity. As the commercially available ligands are unable to effectively probe this region of the cyclophilin family, we turned to in silico techniques to obtain insight into isoform gatekeeper identity and its relationship to accessibility to the -2 pocket. Four hundred test peptides of the general form Xaa-Zaa-Gly-Pro were docked into a subset of cyclophilin family members (PPIA, PPIL2, PPIC, PPWD1, and NKTR) in silico. The isoforms were chosen due to the diversity of the amino acids in the gatekeeper and -2 pocket regions (Figure 5). Monte Carlo simulations were performed to sample conformational space for each combination of cyclophilin isoform and test peptide, allowing flexibility of the tetrapeptide and the sidechains of the gatekeepers at positions comparable to PPIA Thr73 ( gatekeeper 1 ), Lys82 ( gatekeeper 2 ), and Ala103 ( gatekeeper 3 ), and keeping the rest of the receptor rigid [56]. For PPIL2 the sidechain of Arg377 , which substitutes for a glycine in the other cyclophilins investigated, was also allowed flexibility as it contributes a unique chemistry to the -2 region. Throughout the Monte Carlo simulations (200,000 iterations) tethers were imposed on the C-terminal Gly and Pro residues to ensure that the tetrapeptides would remain bound to the active site. For each combination of cyclophilin isoform and tetrapeptide the lowest energy complex was chosen as the predicted conformation of the bound complex, and an estimate of the binding energy was calculated using ICM [57]. Additionally, since low energy complexes may or may not include significant interactions at the -2 pocket, the distance between the tetrapeptide and the Cα of the gatekeeper equivalent to PPIA Lys82 was calculated. This metric was designed to query for tetrapeptides that both bind with favorable energy in the -2 pocket, and also fill the -2 pocket if possible.
For PPIA, the results of the simulations indicate a general preference for aromatics bound at the -2 position , with a preference for tryptophan over tyrosine and almost no peptides selected for with phenylalanine (Figure 6). In addition, there were a few peptides containing methionine, lysine, or arginine at the -2 position that extended deeply into the -2 pocket, albeit with poor predicted binding energies. Peptides with isoleucine, leucine, valine, proline, alanine, glycine, cysteine, threonine, or serine at the -2 position were disfavored, with poor predicted binding energies. There seems to be much less discrimination for the identity of the -3 position, although there is a clear preference for acidic chemistries over basic ones (Figure 6). Visual inspection of a model complex (PPIA+EWGP) shows that a tryptophan at the -2 position is well able to fill the -2 pocket of PPIA, while inspection of other models (PPIA+EFGP or DYGP) shows incomplete entry into the -2 pocket. In addition, these models indicate that the residue at the -3 position often interacts with the residue at the gatekeeper 1 residue, or extends back towards the proline pocket and the key active site residue Arg55.
For validation of these results, we looked at the available data on specificity for the PPIA isoform. Previous in vitro phage display experiments with PPIA (designed to probe substrate preferences at the +1 to +6 positions) found a strong preference for phenylalanine at the -2 and glutamic acid at the -3 position; these residues were provided by the expression vector used in the phage display and therefore biased the pool of samples available for initial selection [32]. Substitution of this glutamic acid/phenylalanine series with any other residues, however, lessened the signal on an array, thereby confirming a preference for these chemistries in solution. Our simulations support this chemical preference for acidic residues at -3 followed by aromatic residues at -2 positions (Figure 6). Another well-characterized substrate for PPIA is the HIV capsid; there are several sequence variants that have been studied both in solution and in crystallographic experiments, and all sequences have either methionine or alanine at the -2 position and histidine or alanine at the -3 position [45, 58]. In the structures of PPIA with these peptides, the alanine does not fill the -2 pocket , and this is likely the reason why it does not score well in our modeling trials. Neither histidine nor alanine at the -3 position is predicted to score highly by our modeling trials, and in the co-crystal structures these residues are not making any significant contacts to the gatekeeper 1 region of PPIA. The validated in vivo substrate CD147 was also investigated; the natural sequence that is acted upon by PPIA is A-L-W-P, which was not predicted to bind tightly to PPIA based either on the phage display data or our simulations and experimentally was found to have rather weak affinity [16]. Finally, the PPIA substrate Itk contains the targeted sequence E-N-N-P, which is a relatively high scoring -3 and -2 sequence combination based on our models [13]. Our simulations recapitulate the experimental data that is available, but imply that none of the in vitro or in vivo substrates studied to date for PPIA interact with the -2 pocket with optimized space-filling or energetic properties.
Interestingly, for four other PPI domains studied using these computational methods (PPIC, PPIL2, NKTR, and PPWD1), distinct patterns of chemical preference are noted for each isoform (Figure 6). Much like PPIA, the PPI domains of PPIC and PPIL2 showed an energetic preference for tryptophan at the -2 position; and for PPIL2 and NKTR it is clear that isoleucine, leucine, valine, proline, alanine, glycine, cysteine, threonine, and serine at the -2 position result in poor predicted binding energies and little penetration into the -2 pocket (Figure 6). However, both the nature of the residues positively selected for and against was different for each of the isoforms selected for analysis. For NKTR there were relatively few tetrapeptide combinations with either favorable predicted binding energy or penetration into the -2 pocket; this is easily rationalized by the extremely narrow gap between the gatekeeper 1 and gatekeeper 3 regions in the NKTR structure, which occlude the -2 pocket and restrict the types of residues that can stably associate with the pocket without steric or charge clash (Figures 5 and 6). PPIC showed a clear preference for tryptophan at the -2 position - similar to PPIA - but tyrosine, phenylalanine, and methionine at the -2 position have comparable energies to tryptophan. Diverse chemistries, including lysine, arginine and even glycine at the -2 position resulted in favorable binding energies in the PPIC dataset. Interestingly, glutamic acid and aspartic acid were energetically disfavored at both the -2 and -3 positions, contrary to what was observed for PPIA (Figure 6). This is most likely due to the substitution of gatekeeper 2 and the overall acidic character of this region of PPIC relative to PPIA (see Figure 5).
|
|
|
Figure 5. The diverse surfaces of the human cyclophilins. Surfaces of the human cyclophilins are shown colored by qualitative electrostatic potential. The scales of the potentials are all roughly the same (averagepotential: -/+65 kBT/e) and range from -/+ 56 kBT/e for PPIGto -/+ 81kBT/e for PPIL4; all surfaces were calculated using the protein contact potential function in PyMol. As discussed in the text, the surfaces have been generally divided into those with neutral or mixed charge character surrounding the -2 pocket; those with largely acidic character around the -2 pocket; and those whose gatekeeper residue identities lead to occlusion of the -2 pocket. |
In the case of PPIL2, there is near equivalency between the aromatics at position -2, with perhaps a slight energetic preference for tryptophan but strong preferences for tyrosine and phenylalanine as well. Peptides that have methionine, glutamine, asparagine, arginine or lysine at the -2 position were often predicted to bind within the -2 pocket, but with a much less favorable binding energy compared to the peptides with aromatic residues at the equivalent position. Likewise there is little discrimination at the -3 position (Figure 6). Compared to PPIL2 simulations, the results for PPWD1 are striking: the acidic surface characteristics of this isoform select strongly for an arginine at the -2 position, with lysine and aromatic residues also yielding good predicted binding energies (Figures 5 and 6). Of the surfaces tested, only PPWD1 provided a surface where strong energy scores were measured for basic residues at this position. For PPWD1, it is interesting to note that the construct used for crystallization contained a sequence AEGP which is encoded N-terminally to the PPI domain and this sequence was found associated with a neighboring PPI domain in the crystal structure. NMR based assays showed that AEGP was able to bind to PPWD1 but was not a good substrate for the enzyme, which correlates well with the poor binding energy predicted for the AEGP tetrapeptide in our simulations [29]. Again, the scarcity of experimental data for cyclophilin isoforms limits the ability to validate the simulations; but to the extent that such information exists, it correlates well with our in silico findings.
In conclusion, there are cyclophilin family members that while sharing overall conservation with the active members of the family do not seem to possess isomerase activity. All three of these isoforms are multi-domain cyclophilins (Figure S3), but only in the case of PPIL2 is there any function associated with the extra-PPI domain [33]. However, at least for PPIL2 and SDCCAG-10, both of which have been found associated with spliceosomal complexes, it may be that (as in the case of PPIH and PPIL1) it is the non-active surface of the PPI domain that performs the major function for this region of these proteins rather than the active surface described here. Additionally, it may well be that the function of the PPI domain in these cyclophilins is to simply bind proline-containing motifs; our modeling data somewhat suggests that option, as there are many proline-containing peptides that are capable of binding PPIL2 with reasonable binding energy and shape complementarity.
|
|
|
Figure 6. Peptide:protein simulations for five members of the cyclophilin family. (A) Detailed results for PPIA. Simulations were set up with the structure of PPIA as represented in PDB 1AK4 and with 400 peptides corresponding to the sequences X-Z-G-P, where X and Z are each of the possible combinations of thenaturally occurring 20 amino acids. Flexible residues in the protein correspond to the “gatekeepers” and are shown. Below, two graphical representations showing the results of the simulation for PPIA. On the left, a scatter plot with the energy metric on the Y axis and the distance metric on the X axis. The lower left quadrant is where the highest scoring peptide combinations are plotted (greatest negative energy and closest interaction with the -2 pocket). The color of each spot in the plot corresponds to the hydrogen bonding potential between that particular peptide and PPIA, with red indicating greater values and purple indicating lesser values. On the right, the identity of the residue at position -3 is plotted along the X axis, and the identity of the residue at the -2 position is plotted along the Y axis. The general chemical classification for each residue is indicated. At the intersection of each X, Y point is a square representing the binding energy and distance metrics. Red indicates greater binding energy for that X, Y pair; purple indicates lesser energy. Larger squares fill the -2 pocket to a greater extent. (B) X, Y arrays for four other cyclophilin simulations. Coloring and axes are as in (A). |
Assays that detect binding of cyclosporin or tetrapeptide substrates are limited by how little specificity is contributed by the proline binding pocket and the Trp121 residue regions of the isomerase domain. Indeed there seems to be almost no selectivity for either cyclosporin or sanglifehrin by human cyclophilin isoforms; and although a recent report focusing on aryl 1-indanylketones showed binding to PPIA, PPIF, and PPIL1 while not binding to PPIB, PPIC, or PPIH [59], it seems that any ligand that coordinates exclusively with the proline pocket and/or Trp121 region is unlikely to be selective with respect to the entire cyclophilin family. Potentially, the +2/+3 region of the isomerase domain could be a site of selectivity; it is clear from our surface representations (Figure 5) that this is a variable part of the cyclophilin domain. However, our results indicate that a clear virtual chemical fingerprint exists for the -2 and -3 positions of the isomerase domain. For instance, PPIA and PPWD1 seem to have restricted sets of sidechains that are preferred at the -2 position (and the -3 position in the case of PPIA), while PPIC appears to be more promiscuous. The highly occluded nature for the -2 pocket exhibited by NKTR results in a restrictive set of allowed tetrapeptide sequences for this isoform; several other isoforms in the cyclophilin family also exhibit this type of gatekeeper restriction. Due to the very distinct molecular features of the -2 region, both in terms of the highly "druggable" -2 pocket and the chemical diversity seen for the gatekeeper residues, it is likely that targeting this region of the cyclophilins for pharmacophore design and selection is more likely to result in tight binders with greater specificity for particular isoforms in the family.
Acknowledgements
The authors thank Angela Mok for preparation of the ISee package. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Financial Disclosure
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX (1989) Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337: 476-478.
- Galat A, Metcalfe SM (1995) Peptidylproline cis/trans isomerases. Progress in Biophysics & Molecular Biology 63: 67-118.
- Takahashi N, Hayano T, Suzuki M (1989) Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337: 473-475.
- McKeon F (1991) When worlds collide: immunosuppressants meet protein phosphatases. Cell 66: 823-826.
- Schreiber SL (1991) Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251: 283-287.
- Gething MJ, Sambrook J (1992) Protein folding in the cell. Nature 355: 33-45.
- Kimmins S, MacRae TH (2000) Maturation of steroid receptors: an example of functional cooperation among molecular chaperones and their associated proteins. Cell Stress & Chaperones 5: 76-86.
- Baker EK, Colley NJ, Zuker CS (1994) The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. EMBO Journal 13: 4886-4895.
- Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, et al. (2005) Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Molecular Cell 19: 111-122.
- Scarlata S, Carter C (2003) Role of HIV-1 Gag domains in viral assembly. Biochimica et Biophysica Acta 1614: 62-72.
- Lu KP, Liou YC, Zhou XZ (2002) Pinning down proline-directed phosphorylation signaling. Trends in Cell Biology 12: 164-172.
- Lippens G, Landrieu I, Smet C (2007) Molecular mechanisms of the phospho-dependent prolyl cis/trans isomerase Pin1. FEBS Journal 274: 5211-5222.
- Brazin KN, Mallis RJ, Fulton DB, Andreotti AH (2002) Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin A. ProcNatlAcadSciUSA 99: 1899-1904.
- Dorfman T, Weimann A, Borsetti A, Walsh CT, Gottlinger HG (1997) Active-site residues of cyclophilin A are crucial for its incorporation into human immunodeficiency virus type 1 virions. Journal of Virology 71: 7110-7113.
- Yurchenko V, Zybarth G, O'Connor M, Dai WW, Franchin G, et al. (2002) Active site residues of cyclophilin A are crucial for its signaling activity via CD147. Journal of Biological Chemistry 277: 22959-22965.
- Schlegel J, Redzic JS, Porter CC, Yurchenko V, Bukrinsky M, et al. Solution Characterization of the Extracellular Region of CD147 and Its Interaction with Its Enzyme Ligand Cyclophilin A. Journal of Molecular Biology In Press, Corrected Proof.
- Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, et al. (2009) The Isomerase Active Site of Cyclophilin A Is Critical for Hepatitis C Virus Replication. J Biol Chem 284: 16998-17005.
- Reidt U, Wahl MC, Fasshauer D, Horowitz DS, Luhrmann R, et al. (2003) Crystal structure of a complex between human spliceosomal cyclophilin H and a U4/U6 snRNP-60K peptide. Journal of Molecular Biology 331: 45-56.
- Xu C, Zhang J, Huang X, Sun J, Xu Y, et al. (2006) Solution structure of human peptidyl prolyl isomerase-like protein 1 and insights into its interaction with SKIP. Journal of Biological Chemistry 281: 15900-15908.
- Wang Y, Han R, Zhang W, Yuan Y, Zhang X, et al. (2008) Human CyP33 binds specifically to mRNA and binding stimulates PPIase activity of hCyP33. FEBS Letters 582: 835-839.
- Leung AW, Halestrap AP (2008) Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochimica et Biophysica Acta 1777: 946-952.
- Leung AW, Varanyuwatana P, Halestrap AP (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. Journal of Biological Chemistry 283: 26312-26323.
- Dubourg B, Kamphausen T, Weiwad M, Jahreis G, Feunteun J, et al. (2004) The human nuclear SRcyp is a cell cycle-regulated cyclophilin. Journal of Biological Chemistry 279: 22322-22330.
- Teigelkamp S, Achsel T, Mundt C, Gothel SF, Cronshagen U, et al. (1998) The 20kD protein of human [U4/U6.U5] tri-snRNPs is a novel cyclophilin that forms a complex with the U4/U6-specific 60kD and 90kD proteins. RNA 4: 127-141.
- Anderson SK, Gallinger S, Roder J, Frey J, Young HA, et al. (1993) A cyclophilin-related protein involved in the function of natural killer cells. ProcNatlAcadSciUSA 90: 542-546.
- Schonbrunner ER, Mayer S, Tropschug M, Fischer G, Takahashi N, et al. (1991) Catalysis of protein folding by cyclophilins from different species. Journal of Biological Chemistry 266: 3630-3635.
- Price ER, Zydowsky LD, Jin MJ, Baker CH, McKeon FD, et al. (1991) Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proceedings of the National Academy of Sciences of the United States of America 88: 1903-1907.
- Friedman J, Weissman I (1991) Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell 66: 799-806.
- Davis TL, Walker JR, Ouyang H, MacKenzie F, Butler-Cole C, et al. (2008) The crystal structure of human WD40 repeat-containing peptidylprolyl isomerase (PPWD1). Febs J 275: 2283-2295.
- Hoffmann K, Kakalis LT, Anderson KS, Armitage IM, Handschumacher RE (1995) Expression of human cyclophilin-40 and the effect of the His141-->Trp mutation on catalysis and cyclosporin A binding. European Journal of Biochemistry 229: 188-193.
- Mi H, Kops O, Zimmermann E, Jaschke A, Tropschug M (1996) A nuclear RNA-binding cyclophilin in human T cells. FEBS Letters 398: 201-205.
- Piotukh K, Gu W, Kofler M, Labudde D, Helms V, et al. (2005) Cyclophilin A binds to linear peptide motifs containing a consensus that is present in many human proteins. Journal of Biological Chemistry 280: 23668-23674.
- Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI (2001) U box proteins as a new family of ubiquitin-protein ligases. Journal of Biological Chemistry 276: 33111-33120.
- Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, et al. (2007) A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proceedings of the National Academy of Sciences of the United States of America 104: 20523-20528.
- Senisterra GA, Markin E, Yamazaki K, Hui R, Vedadi M, et al. (2006) Screening for ligands using a generic and high-throughput light-scattering-based assay. Journal of Biomolecular Screening 11: 940-948.
- Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr., et al. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proceedings of the National Academy of Sciences of the United States of America 103: 15835-15840.
- Cavarec L, Kamphausen T, Dubourg B, Callebaut I, Lemeunier F, et al. (2002) Identification and characterization of Moca-cyp - A Drosophilia melanogaster nuclear cyclophilin. Journal of Biological Chemistry 277: 41171-41182.
- Kern D, Kern G, Scherer G, Fischer G, Drakenberg T (1995) Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry 34: 13594-13602.
- Harrison RK, Stein RL (1990) Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry 29: 3813-3816.
- Pirkl F, Buchner J (2001) Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. Journal of Molecular Biology 308: 795-806.
- Dartigalongue C, Raina S (1998) A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO Journal 17: 3968-3980.
- Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols 4: 363-371.
- Ke H, Mayrose D, Belshaw PJ, Alberg DG, Schreiber SL, et al. (1994) Crystal structures of cyclophilin A complexed with cyclosporin A and N-methyl-4-[(E)-2-butenyl]-4,4-dimethylthreonine cyclosporin A. Structure 2: 33-44.
- Zhao Y, Chen Y, Schutkowski M, Fischer G, Ke H (1997) Cyclophilin A complexed with a fragment of HIV-1 gag protein: insights into HIV-1 infectious activity. Structure 5: 139-146.
- Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP (2003) Structural insights into the catalytic mechanism of cyclophilin A. Nature Structural Biology 10: 475-481.
- Bossard MJ, Koser PL, Brandt M, Bergsma DJ, Levy MA (1991) A single Trp121 to Ala121 mutation in human cyclophilin alters cyclosporin A affinity and peptidyl-prolyl isomerase activity. Biochemical & Biophysical Research Communications 176: 1142-1148.
- Liu J, Chen CM, Walsh CT (1991) Human and Escherichia-Coli Cyclophilins - Sensitivity to Inhibition by the Immunosuppressant Cyclosporine-A Correlates with A Specific Tryptophan Residue. Biochemistry 30: 2306-2310.
- Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, et al. (1992) Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Science 1: 1092-1099.
- Kajitani K, Fujihashi M, Kobayashi Y, Shimizu S, Tsujimoto Y, et al. (2008) Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-A resolution. Proteins 70: 1635-1639.
- Mark P, Nilsson L (2007) A molecular dynamics study of Cyclophilin A free and in complex with the Ala-Pro dipeptide. European Biophysics Journal 36: 213-224.
- Leone V, Lattanzi G, Molteni C, Carloni P (2009) Mechanism of action of cyclophilin a explored by metadynamics simulations. PLoS Computational Biology 5: e1000309.
- Galat A (1999) Variations of sequences and amino acid compositions of proteins that sustain their biological functions: An analysis of the cyclophilin family of proteins. Archives of Biochemistry & Biophysics 371: 149-162.
- Eisenmesser EZ, Bosco DA, Akke M, Kern D (2002) Enzyme dynamics during catalysis.[see comment]. Science 295: 1520-1523.
- Scholz C, Rahfeld J, Fischer G, Schmid FX (1997) Catalysis of protein folding by parvulin. Journal of Molecular Biology 273: 752-762.
- Satish Babu M, Per H, Uno C (2009) A nonessential role for Arg 55 in cyclophilin18 for catalysis of proline isomerization during protein folding. Protein Science 18: 475-479.
- Abagyan R, Totrov M (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. Journal of Molecular Biology 235: 983-1002.
- Schapira M, Totrov M, Abagyan R (1999) Prediction of the binding energy for small molecules, peptides and proteins. Journal of Molecular Recognition 12: 177-190.
- Vajdos FF, Yoo S, Houseweart M, Sundquist WI, Hill CP (1997) Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci 6: 2297-2307.
- Daum S, Schumann M, Mathea S, AumuÌ?ller T, Balsley MA, et al. (2009) Isoform-Specific Inhibition of Cyclophilins. Biochemistry 48: 6268-6277.
- Minor W, Cymborowski M, Otwinowski Z (2002) Automatic system for crystallographic data collection and analysis. Acta Physica Polonica A 101: 613-619.
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276: 307-326.
- Read RJ (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta CrystallogrDBiolCrystallogr 57: 1373-1382.
- Collaborative Computational Project N (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallographica Section D 50: 760-763.
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallographica - Section a - Foundations of Crystallography 47: 110-119.
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography 60: 2126-2132.
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta CrystallogrDBiolCrystallogr 57: 122-133.
- Potterton E, Briggs P, Turkenburg M, Dodson E (2003) A graphical user interface to the CCP4 program suite. Acta Crystallographica Section D-Biological Crystallography 59: 1131-1137.
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nature Structural Biology 6: 458-463.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography 26: 283-291.
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Research 35: W375-383.
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, et al. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes.[see comment]. Journal of Biomolecular NMR 6: 277-293.
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, et al. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687-696.
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, et al. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Research 33: W299-302.
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends in Biochemical Sciences 23: 403-405.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876-4882.
- Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127-128.
Download Standalone iSee datapack: You can download and view all the Information of a datapack offline including information not available in the web version (where applicable). You will also need to download and install the ICM-Browser to view the standalone datapacks.


Datapack created using Molsoft ICM and Molsoft Browser technologies. (Patent Pending)