|
|

The Human Cyclophilin Family of Peptidyl-Prolyl Isomerases - Davis et al.
|
|
|
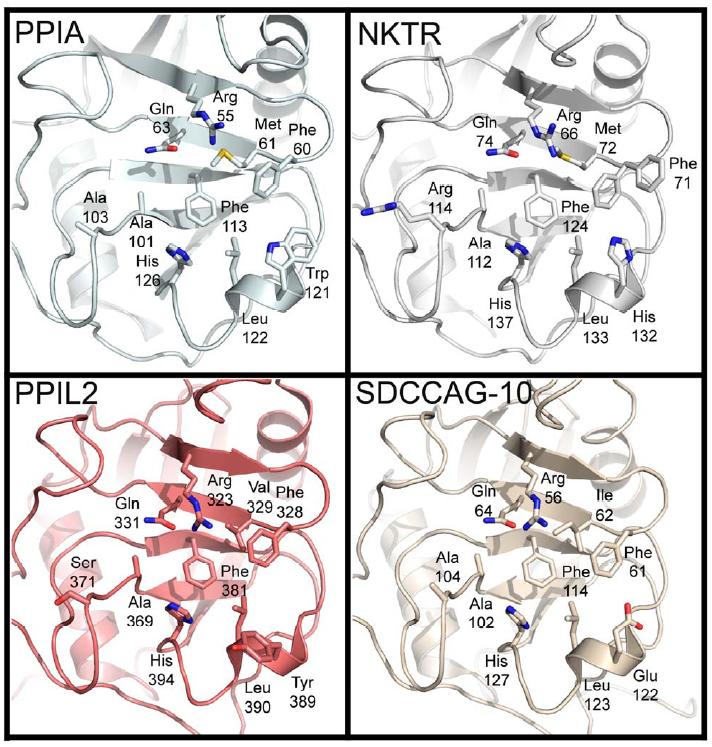
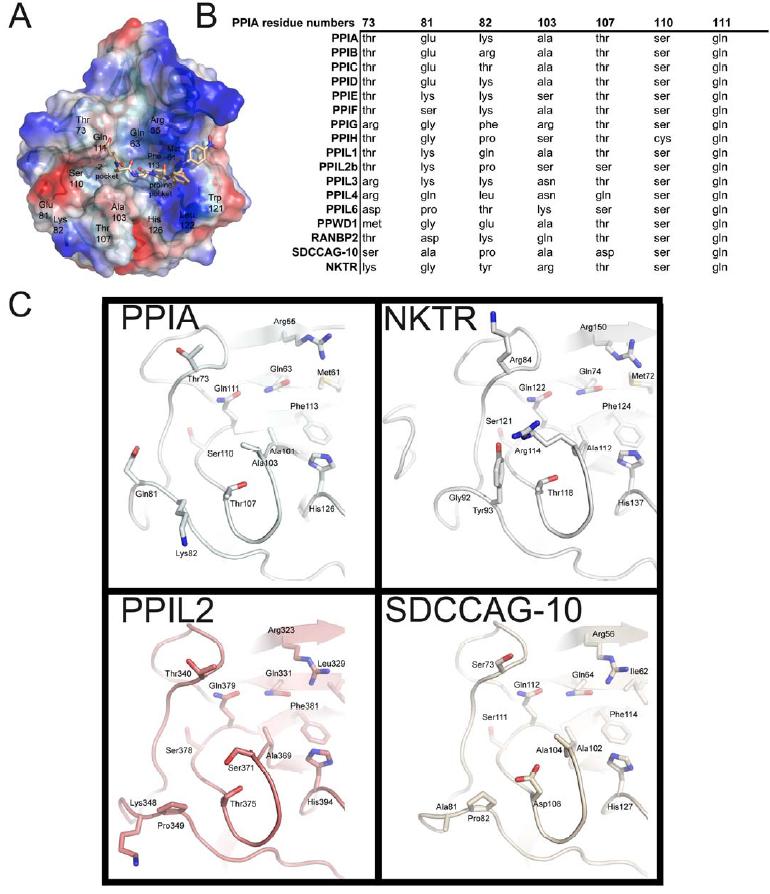
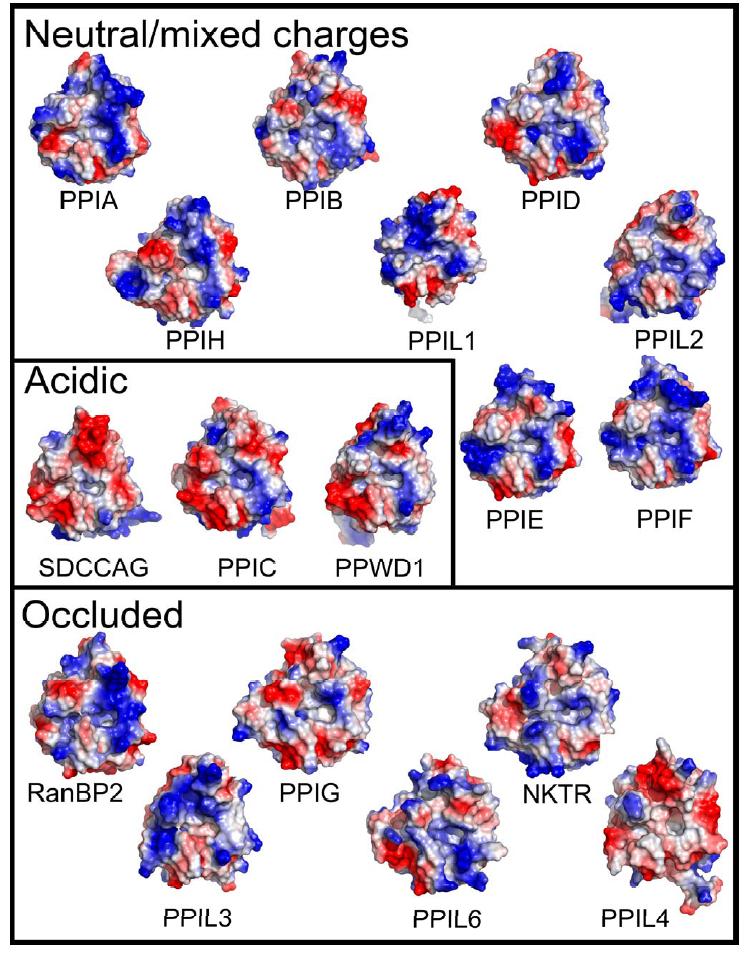
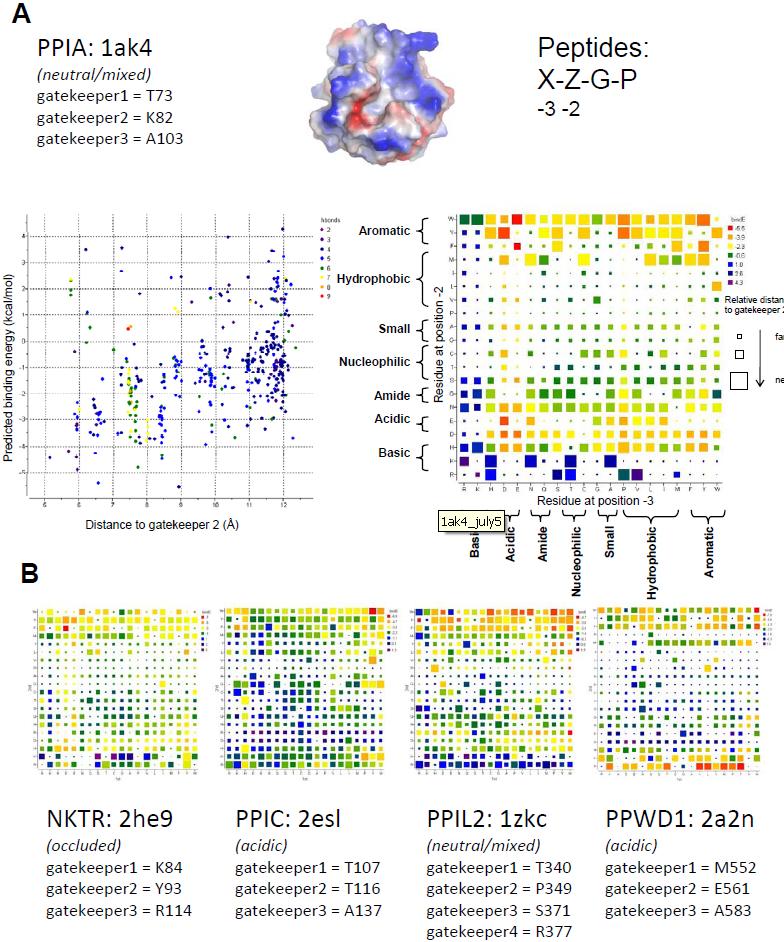
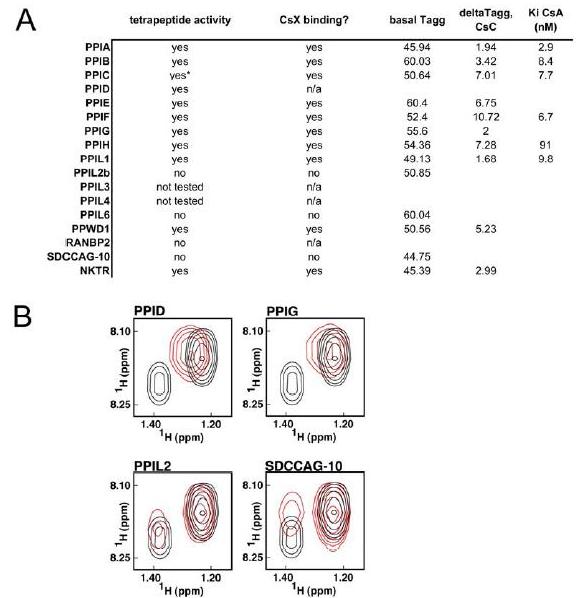
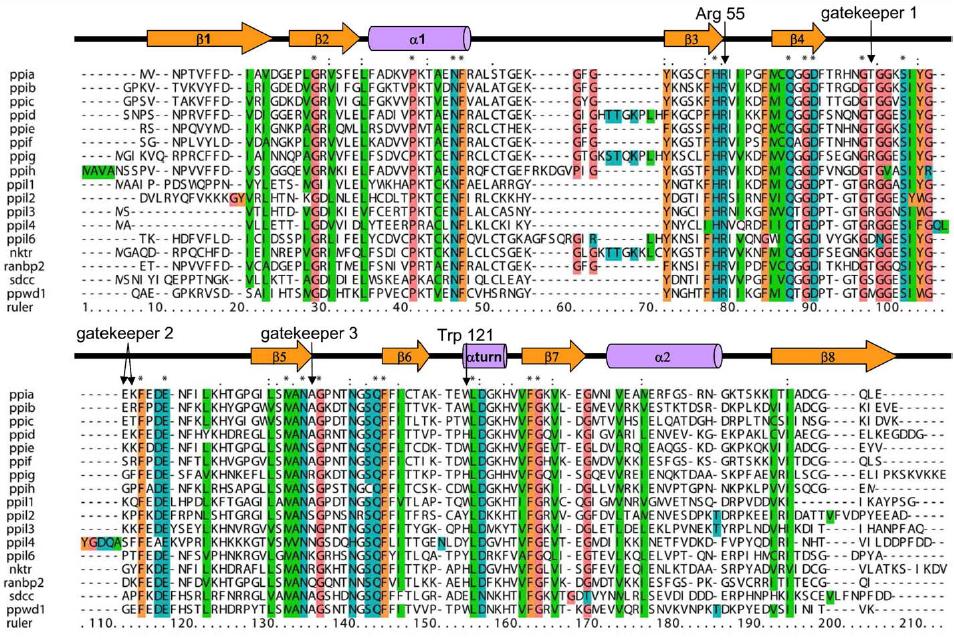
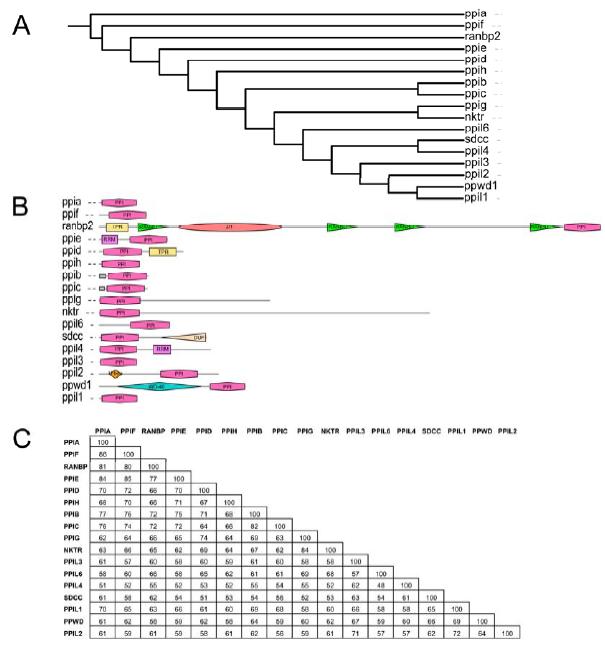
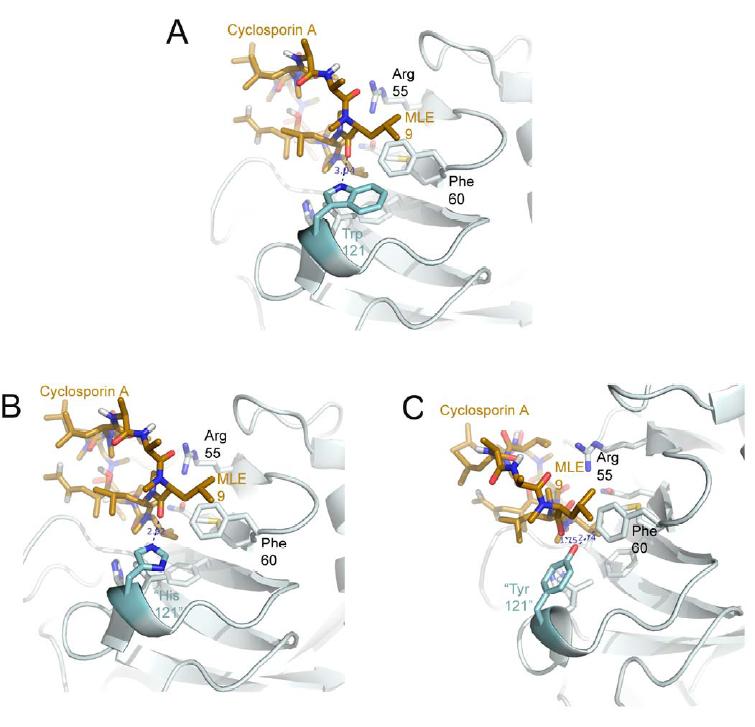
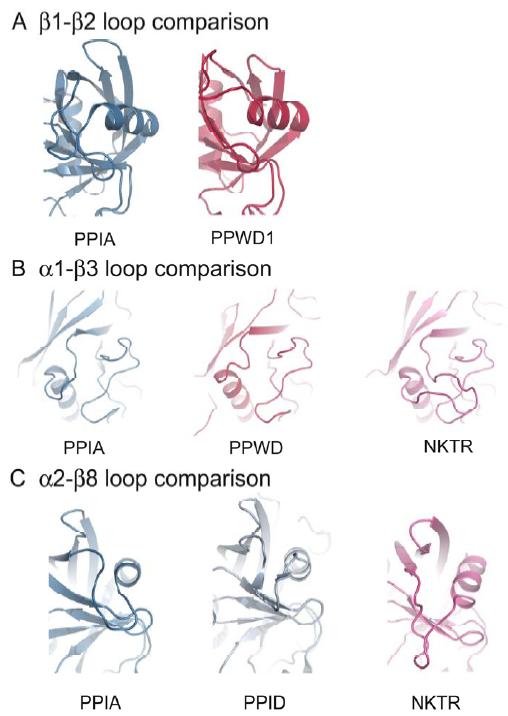
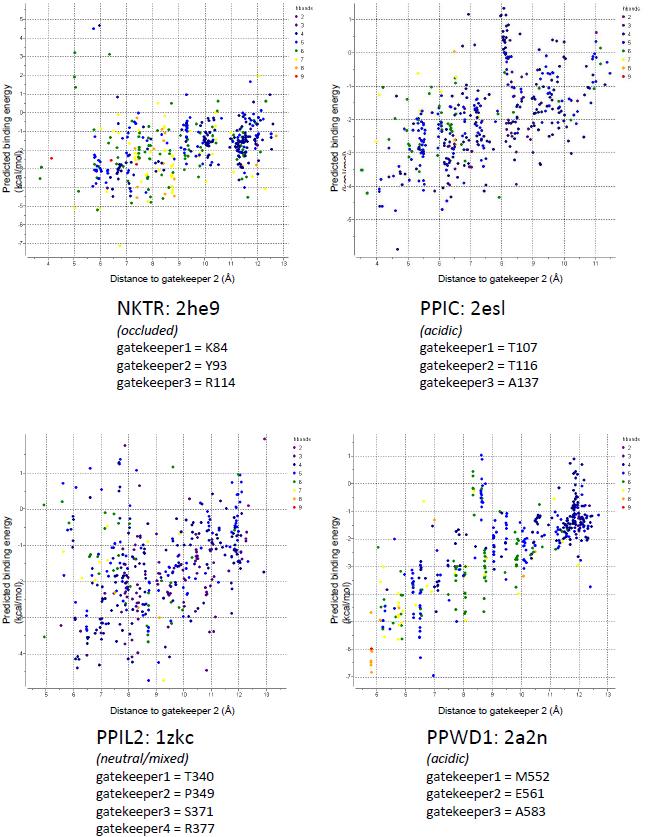
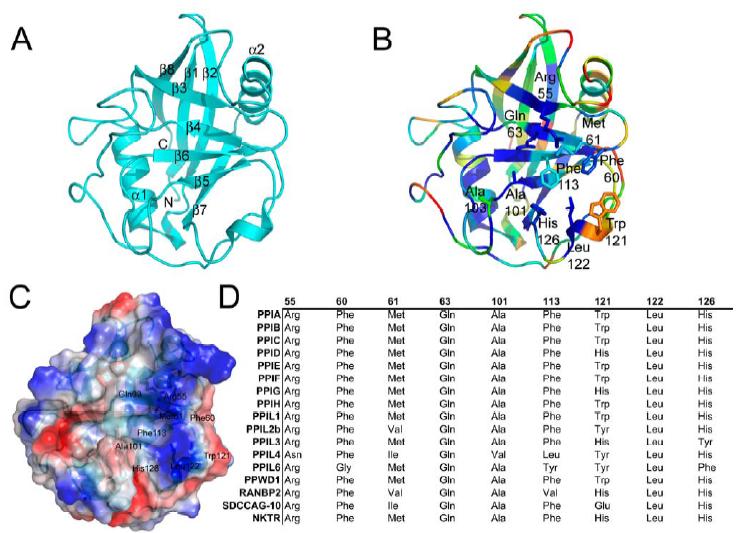
Figure 1. Structural elements of the cyclophilin fold and the definition of the active surface of PPIA. (A) Secondary structural elements of PPIA in ribbon representation, with key structural elements labeled. All structural outputs were generated using PyMol unless otherwise noted. (B) Consurf representation of sequence conservation within the human cyclophilin family; residues that comprise the active surface of the cyclophilin family are labeled [73]. (C) The molecular surface of the PPIA structure. The view is oriented roughly as in (A) and (B). (D) Comparison of the sequences that define the active surface of the PPI domain. Residue numbering corresponds to PPIA. |
Download Standalone iSee datapack: You can download and view all the Information of a datapack offline including information not available in the web version (where applicable). You will also need to download and install the ICM-Browser to view the standalone datapacks.


Datapack created using Molsoft ICM and Molsoft Browser technologies. (Patent Pending)