|
|
1. Structural Genomics Consortium, University of Toronto, MaRS center, South Tower, 7th floor, 101 College Street, Toronto, Ontario, Canada, M5G 1L7. 2. Ontario Cancer Institute and Department of Medical Biophysics, University of Toronto, Canada. 3. Current address: Department of Structural and Chemical Biology, Mount Sinai School of Medicine, One Gustave L. Levy Place, Icahn Medical Institute, Box 1677, New York, NY 10029, USA. 4. Department of Pharmacology and Toxicology, University of Toronto, Medical Sciences Building, 1 King's College Circle, Toronto, Ontario, Canada M5S 1A8.
Funding: This work is supported by the Structural Genomics Consortium (SGC), a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust.
SET domain methyltransferases deposit methyl marks on specific histone tail lysine residues and play a major role in epigenetic regulation of gene transcription. We solved the structures of the catalytic domains of GLP, G9a, Suv39H2 and PRDM2, four of the eight known human H3K9 methyltransferases in their apo conformation or in complex with the methyl donating cofactor, and peptide substrates. We analyzed the structural determinants for methylation state specificity, and designed a G9a mutant able to tri-methylate H3K9. We show that the I-SET domain acts as a rigid docking platform, while induced-fit of the Post-SET domain is necessary to achieve a catalytically competent conformation. We also propose a model where long-range electrostatics bring enzyme and histone substrate together, while the presence of an arginine upstream of the target lysine is critical for binding and specificity.
Introduction
Post-translational modifications of histone proteins regulate chromatin compaction, mediate epigenetic regulation of transcription, and control cellular differentiation in health and disease [1,2]. Methylation of histone tails is one of the fundamental events of epigenetic signaling [3]. Tri-methylation of lysine 9 of histone 3 (H3K9) mediates chromatin recruitment of HP1, heterochromatin condensation and gene silencing [4,5]. Similarly, methylation of H3K27 and H4K20 are associated with a repressed state of chromatin, whereas expressed genes are methylated at H3K4, H3K36 and H3K79 ([3,6] for review). Histone methyltransferases are divided into protein arginine methyltransferases (PRMTs) and histone lysine methyltransferases (HKMTs). HKMTs catalyze the transfer of a methyl group from the co-factor S-adenosyl-L-methionine (SAM) to a substrate lysine and, with the exception of DOT1L, are all organized around a canonical SET domain [7,8]. The structures of a number of HKMTs have been reported, including ternary complexes of human orthologs with co-factor and substrate peptides (SETD7-H3K4, SETD8-H4K20 and MLL1-H3K4 [9,10,11,12]), as well as N. crassa Dim-5 in complex with a H3K9 peptide [13] and a viral protein complexed to H3K27 [14] (Figure 1). These structures collectively highlighted a remarkable plasticity of the peptide-binding site and a lack of clear structural motifs that correlate with sequence selectivity [8,15].
Methylation of H3K9 in humans relies mostly on members of the Suv39 family, namely EHMT1/GLP, EHMT2/G9a, SUV39H1, SUV39H2, SETDB1 and SETDB2, as well as then non-Suv39 enzymes PRDM2 and ASH1L [6] (Figure1). Here we report the high-resolution crystal structures of the methyltransferase domains of GLP, G9a, SUV39H2 and PRDM2, and propose a structural mechanism underlying substrate recognition. Our data also provide important insight to guide the development of potent and selective inhibitors of HKMTs, which are likely to have applications in a variety of diseases including regenerative medicine, oncology and inflammation [16,17,18,19,20] ([21,22] for review).
|
|
|
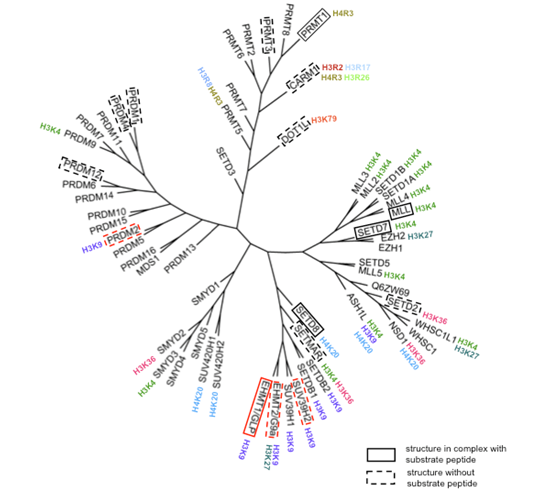
Figure 1: Phylogenetic tree of human histone methyltransferases. Phylogeny is based on a multiple sequence alignment of the methyltransferase domain including the N-SET, Pre_SET, SET, I-SET, and Post-SET motifs. Substrate selectivity was extracted from Kouzarides [1]. Enzymes with solved structure are highlighted by a frame, dotted if no peptide substrate complex is available. Structures solved in the present work are framed in red. |
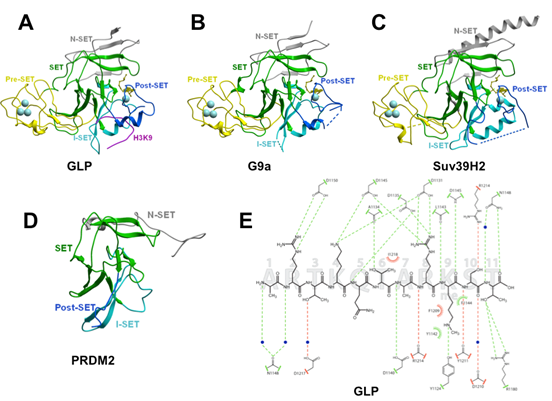
Overall structure: We have solved the crystal structures of the catalytic domain of four H3K9 methyltransferases: (1) the complexes of GLP/EHMT1 co-crystallized with the co-factor product S-adenosyl-L-homocysteine (SAH) alone and with an H3K9me or H3K9me2 peptide, (2) G9a/EHMT2 and SUV39H2 in complex with SAH and SAM respectively, and (3) the non Suv39 protein PRDM2. The Suv39 structures
G9a,
GLP and
SUV39H2 adopt a typical fold composed of a conserved SET domain and variable I-SET insert, flanked by Pre- and Post-SET regions, and characterized by canonical features such as a
pseudo-knot next to the catalytic site, distinct co-factor and substrate binding areas meeting at the
site of methyl transfer, and a narrow substrate
lysine docking channel [8,23,24]. The Pre-Set domain is absent in PRDM2, where N-SET and SET domains are fused (Figure 2D). The
four structures are also characterized by the presence of an N-SET region located N-terminal to the Pre-SET, that wraps around the core SET domain (Figure 2A-D). The H3K9 peptide
lies in a groove formed by the I-SET and Post-SET domains (Figure 2A), as previously observed in other HKMTs ([8] for review), and makes extensive contact with the enzyme through both backbone and side-chain interactions (Figure 2E).
|
|
|
Figure 2: Structures of four human H3K9 HKMTs. (A) a ternary complex of GLP with SAH and a H3K9Me substrate peptide, (B) G9a in complex with SAH, (C) SUV39H2 in complex with SAM and (D) PRDM2, highlighting Pre-Set, SET, I-SET, Post-SET domains and the conserved presence of an N-SET domain. The co-factor is shown as yellow sticks. Residues flanking un-resolved regions are connected by dotted lines. (E) The detail of the interactions between GLP and an H3K9Me peptide. |
(TIP 1: the background colour can be changed to [WHITE] or [BLACK])
(TIP 2: reset the view)
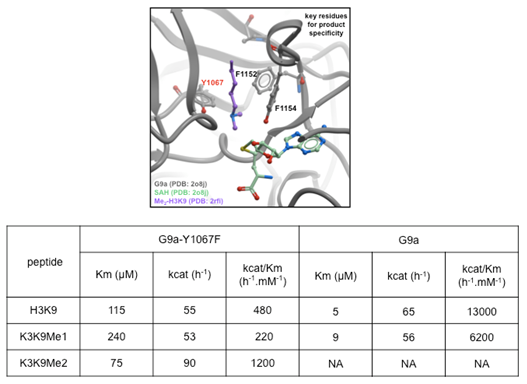
Methylation state specificity: Mono-, di- or tri- methylation of H3K9 constitute distinct biochemical signals and are established by distinct histone methyltransferases; G9a and GLP are mono- and di-methylases, and SUV39H2 di- and tri-methylates a mono-methylated substrate. The specificity of PRDM2 is unknown [25]. Several aromatic residues line the GLP channel occupied by the substrate lysine leading to the catalytic site and contribute to methylation state specificity as previously noted for several other HKMTs [9,12,26,27]. Two residues are of particular importance for catalysis. First, a
conserved tyrosine residue of the post-SET domain (Y1211 in GLP and Y1154 in G9a) is a major component of the lysine binding channel while its hydroxyl group participates in catalysis. This residue cannot be mutated without losing catalytic activity (Figure 3) [8,28]. Second, the hydroxyl group of GLP's Y1124 (Y1067 in G9a) hydrogen-bonds to the methyl-accepting nitrogen, thereby inhibiting an orientation of the dimethyl-amine that would favor transfer of a methyl from SAM, as was previously shown for SETD7 ([12], [28] for review). To confirm this model, we showed that, unlike wild-type G9a, the Y1067F mutant is able to tri-methylate H3K9 (Figure 3). Similarly, it was shown that the F1152Y G9a mutant can only mono-methylate H3K9 [29]. Our structures show that this residue is perfectly superimposed with GLP's F1209 (0.1 Å RMSD), which, if mutated to Tyr, would hydrogen-bond with the ε-amine nitrogen of H3K9 and impair the alignment of the accepting amine's lone pair with the methyl-sulfur bond of SAM (Figure 3). Thus, the methylation state selectivity appears to be inversely proportional to the number of tyrosine residues surrounding the methyl accepting nitrogen.
|
|
|
Figure 3: Structural determinants of G9a mono/di-methylation specificity. A model of substrate lysine-bound G9a (Top panel) was generated from the superposition of the active sites of the GPL ternary complex and GLP. Y1067 of G9a stabilizes the di-methylamine end of the substrate lysine in an orientation where the lone-pair is not facing the co-factor, thereby disfavoring transfer of a third methyl group. The Y1067F mutant loses this restriction and can tri-methylate its substrate. Previous work had shown that the F1152Y G9a mutant can only mono-methylate H3K9 [29]. |
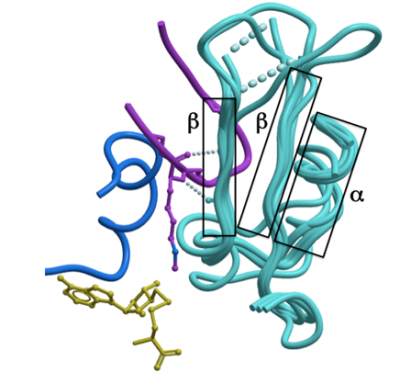
I-SET is a rigid peptide-docking platform: The
structure of the I-SET domain appears relatively conserved, whether in the apo, co-factor-, or peptide-bound form, and is composed of a helix followed by a two-stranded anti-parallel β-sheet, linked by loops of variable lengths (Figure 4). Superimposition of our six new H3K9 methyltransferase structures (see Materials and Methods) shows that in all cases the first β-strand is in a conformation which would preserve the pair of backbone hydrogen-bonds observed between Lys-9 substrate and strand-1 of I-SET in the GLP-peptide complex (Figure 4).
|
|
|
Figure 4: The I-SET domain is relatively rigid and structurally conserved. Structural superimposition of the ternary GLP structure with G9a or Suv39H2 in complex with co-factor and with the apo-structure of PRDM2 shows that the I-SET (cyan) conformation is conserved. The backbone atoms engaged in a double hydrogen-bond with the substrate lysine observed in all available HKMT-peptide complexes are already positioned in the absence of peptide or co-factor. |
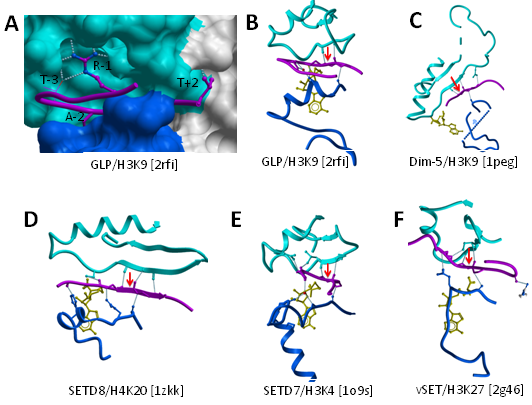
Furthermore, a systematic comparison of the ternary structures presented here for GLP-H3K9 and previously published for Dim-5-H3K9, SETD8-H4K20, SETD7-H3K4, SETD7-TAF10, SETD7-P53, vSET-H3K27 reveals that this pair of hydrogen bonds between the backbone of a single substrate peptide residue and the first strand of I-SET is observed (1) in all HKMT ternary structures to date and (2) always and only at the substrate lysine (Figure 5B-F - SETD7-TAF10 and SETD7-P53 complexes not shown). This suggests that an evolutionary pressure enforces conservation of this "double hydrogen-bond", which likely plays an important role in the binding mechanism, probably by imposing the proper orientation of the peptide when lysine inserts into the active site; flipping the substrate by 180° in its groove would not allow formation of the double hydrogen bond.
|
|
|
Figure 5: Backbone and side-chain contributions to peptide binding. A: the substrate peptide sits in a groove formed by the I-SET (cyan) and the Post-SET (blue) domains. Peptide side-chains contributing most to the interaction are shown (magenta sticks). The guanidinium group of H3R8 (R-1) makes extensive contacts with the I-SET domain. B-F: Both Post-SET (blue) and I-SET (cyan) backbone atoms are engaged in a network of hydrogen-bonds with the peptide main-chain (magenta). A pair of hydrogen-bonds between backbone atoms of the I-SET and substrate lysine are conserved in all available HKMTs ternary complexes to date (dotted lines flanking red arrow). |
Our structures single-out residue R-1 of H3 (the methylated lysine is used as reference position 0 for peptide residue numbering throughout the text) as the major contributor to the interaction after the substrate lysine itself, with four direct hydrogen-bonds between the arginine guanidinium group and GLP (Figure 2E). This is in agreement with recent mutational analysis showing that no substitution at position -1 is tolerated by G9a [30]. This critical interaction takes place exclusively with I-SET residues (Figure 5A). GLP residues that contribute to substrate binding are conserved in G9a, and it is reasonable to assume that the peptide binding mode observed in GLP holds for G9a. On the other hand, mutation of R-1 to alanine only mildly affects peptide binding to Dim-5, a H3K9 methyltransferase in N. crassa [31], indicating that the selectivity mechanism observed for human GLP and G9a is not universal.
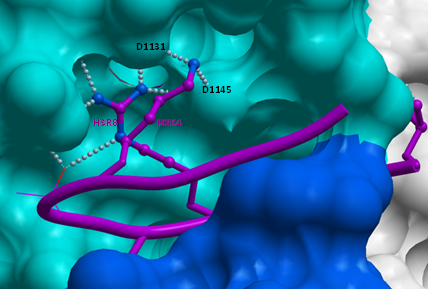
Intriguingly, our structures of GLP in complex with mono- or di-methylated susbtrate peptides reveal that residue H3K4 is making 2 hydrogen-bonds with the D1131 and D1145 side-chains of the I-SET domain (Figure 6), which suggests that H3K4 methylation may lower binding affinity, and reduce H3K9 methylation efficiency. We tested this hypothesis, and observed a mild decrease of 43% in affinity of GLP for a H3K9 peptide trimethylated at lysine 4, and similar reduction in enzymatic efficiency, while Kcat was unaffected. Mono and di-methylation of H3K4 had no or very limited effect (data not shown). It is not clear whether this variation is biologically significant.
|
|
|
Figure 6: Contribution of H3K4 to H3K9 binding. Our structures of GLP in complex with H3K9me or H3K9me2 show that H3K4 folds on top of H3R8, making polar interactions with D1131 and D1145 of GLP. |
These results suggest a model in which a mostly pre-formed I-SET domain acts as a receiving platform for the histone 3 tail. Binding includes a conserved pair of hydrogen-bonds with the backbone of the substrate lysine, and critical contacts with a basic side-chain upstream of the methyl acceptor.
Mobile Post-SET domain closes onto the peptide substrate:The Post-SET domains of G9a, GLP, SUV39H2, but not PRDM2 include a ZnCys motif previously observed in the structures of Dim-5 [13] and the H3K4 methyltransferase MLL1 [10]. The Post-SET domains of G9a and GLP present an α-helix that contributes to peptide binding where other HKMTs have a loop. Unlike I-SET, Post-SET is absent from the PRDM2 structure, which lacks co-crystallized SAM or SAH (Figure 2D). It is partially folded in the structures of G9a and SUV39H2 in complex with SAH and SAM respectively (Figure 2B-C), and fully ordered in the ternary complexes of GLP with SAH and H3K9 peptide (Figure 2A). As previously observed with other HKMTs ([9,11], [8] for review), the co-factor contributes to the formation of a hydrophobic, mostly aromatic cluster (composed of Post-SET Y1211/Y1154/Y261, F1215/F1158, W1216/W1159/L298, F1223/F1166/T285 and SET H1170/H1113/H220 in GLP/G9a/SUV39H2) necessary for partial folding of the Post-SET domain.
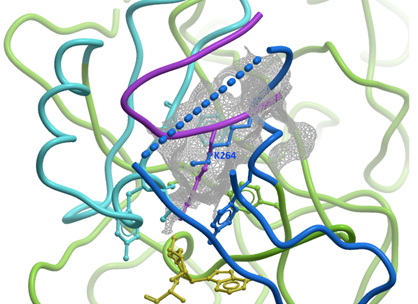
Surprisingly, in our structure of SUV39H2, Post-SET Lys-264 is inserted into the partially formed substrate lysine binding channel, which may represent some form of auto-inhibitory mechanism (Figure 7). Post-SET is fully structured only when bound to the substrate peptide (Figure 2A), or to a small molecule inhibitor [30], but the density is incomplete otherwise (Figure 2B-D). This implies that Post-SET is naturally flexible, which may be important for peptide turn-over, as recently proposed [10].
|
|
|
Figure 7: Auto-inhibitory conformation of SUV39H2. In our structure of SUV39H2, the C-terminus of the Post-Set domain (blue) adopts a conformation that positions its K264 side-chain (blue sticks) half-way into the substrate lysine channel (gray mesh). The H3K9me peptide (magenta) from a superimposed GLP-H3K9me structure is shown as a reference. SET and Post-SET of SUV39H2 are colored green and blue respectively. |
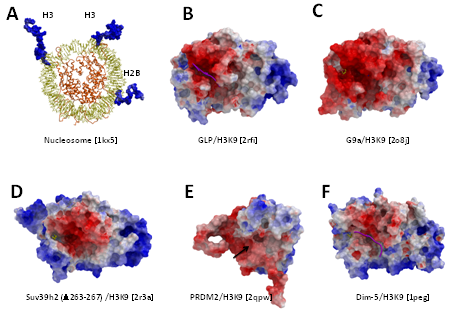
Long range electrostatics attract histone peptides to the HKMT binding groove: Histone tails are rich in lysines and arginines and consequently are electropositive (Figure 8). Mapping the electrostatic potential along the molecular surface of GLP, G9a, SUV39H2 and PRDM2 click here shows that the peptide-binding groove is consistently electronegative (Figure 5B-E). This feature is also conserved in the structure of the N. crassa H3K9 methyltransferase Dim-5 (Figure 5F). This suggests that non-specific long-range electrostatic attractions play an evolutionarily conserved role in guiding the substrate-binding groove towards histone tails.
|
|
|
Figure 8: Electrostatic component to H3K9 peptide binding. While the overall electrostatic profile of available H3K9 methyltransferases structures varies, the peptide-binding groove is consistently electronegative (B-E: this work, F: N. crassa methyltransferase Dim-5), in contrast with the largely positive electrostatic potential of histone tails (A). When present, the substrate peptide is shown in magenta. Residues 264-267 of SUV39H2 were partially occupying the binding site and were removed. The Post-SET domain of PRDM2 is entirely disordered and the position of the substrate lysine binding channel is indicated with a black arrow. |
Based on the four HKMT structures presented here, we propose a mechanism for selective lysine H3K9 methylation, in which (1) long-range electrostatics attract the enzyme onto basic histone tails, (2) a pre-formed I-SET domain carries structural determinants necessary for specific interactions with the substrate peptide, and (3) catalytically competent conformation is achieved by subsequent closing of the Post-SET domain on the substrate. Considering the electronegative potential of the binding groove, our analysis suggests that HKMT inhibitors should be rather basic. To achieve selectivity, inhibitors should bind sites with clear interaction field potential occupied by residues distal to the substrate lysine. A recent co-crystal structure of the first specific HKMT inhibitor supports these general concepts [32].
We thank Peter Loppnau, He Ren, and Yong Zhao for their excellent technical support, and Stephen Frye for comments on the manuscript.