[ VLS Overview ]
by Max Totrov and Ruben Abagyan[ Receptor | Choosing ligands | Docking timing | Scoring | Docking intro | Project setup | _dockBatch | Converting chemicals | Running dock job | VLS Introduction | Vls threshold | Mf score | Admet selection | Parallelization | Vls cluster | Vls scores storage | Make Hitlist | DockScan | Template docking | SLURM SGE | Ligand AIDE | GINGER | RIDGE | CombiRIDGE | V-SYNTHES | GigaScreen ]
This section concerns with predictions of interactions of drugs or small biological substrates (less than about 600-700 dalton) to pockets of larger, more rigid, receptors (typically, protein molecules, DNA or RNA). There are five major steps in docking and screening.
Receptor from PDB
If you have only a single entry with your receptor, convert the protein with
convertObject yes yes no no , after deleting water molecules and irrelevant chains
(e.g. delete a_!1 ), or use menus as in the ligand docking section.
However, if you have a choice between several templates, take the following into account:
- X-ray structure is preferable to an NMR structure
- high resolution X-ray structure ( less than 2.1A ) is much better than, say 2.5A .
- watch out for high-B-factor regions and avoid them; sometimes crystallographers deposit fantasy coordinates with high-B-factors. Use:
orcolor a_//* Bfactor( a_//* ) # from command lineColor/B-factorfrom the Gui-menu . - place polar hydrogens and choose correct form of
histidine(convertObject yes yes no notakes care of that ) - a bound conformation of the receptor is preferable, however if you use an apo-model, an NMR structure or a model by homology,
the side-chains in a pocket may be incorrect. Frequently they stick out and prevent a ligand from binding.
Those stubborn side-chains can be 'tamed', (i) manually; (ii) by a side chain simulation with elevated
surfaceTension;or (iii) by an explicit flexible docking calculation with a known ligand.
Receptor from homology modeling
A model by homology can be built with the build model command (menu
Homology/Build_Model)
followed macro refineModel .
Identifying pockets
If a binding pocket is not known in advance, use
icmPocketFinder or icmCavityFinder (for closed pockets) macros.
icmPocketFinder can also be accessed from menu Docking/Receptor Setup , submenu Identify_Binding_Sites
Ligand from PDB
Then to dock a ligand from pdb, go through the procedure described in the ligand docking section.
Ligand(s) from a mol/mol2- file, or SMILES strings.
The main prerequisite is that the formal charges and the bond types are correct.
If they are not correct, you need to process each molecule manually as described in the ligand docking section.
From a command line you may use the build smiles or convert2Dto3D macro.
Docking menu ).
Some facts about ICM docking:
- an average docking time is 2-30 seconds per ligand per processor
- ICM docking performed very well in predicting the binding geometry in several comparative benchmarks.
- the time per ligand was chosen to be the smallest possible to allow screening of very large data sets.
To increase the time spent per ligand, change the
Docking_effortparameter from theDocking.Small Set Docking Batchmenu to 3. or 5., or supply this parameter to therundockscript directly.
Pitfalls. Inaccurate receptor model, or incorrectly converted ligands, or insufficient optimization effort may lead to incorrect predictions.
learn command).
The vls module allows you to access a good scoring function.
Docking/New Project ).
Avoid spaces and leading digits in the name. All files related to the docking project will be
stored under names, which start from the project name. Most customized parameters will be
saved in the table file under the project name dockProjName.dtb as well:
DOCK1.dtb # control table
DOCK1_rec.ob # receptor object
DOCK1_gb.map # 3D potential grids, or 'maps'
DOCK1_gc.map
DOCK1_ge.map
DOCK1_gh.map
DOCK1_gl.map
DOCK1_gs.map
DOCK1_probe.ob # 4 atom probe for initial superposition (or)
DOCK1_tmplt.ob # template ligand (optional)The next step is to set up the receptor (GUI menu
Docking/New Project ). Select the receptor
molecules, in most cases a_* will do - all molecules in the current object will be included.
Define binding site residues, either manually e.g. a_/123,144,152 for selection by residue
numbers, or graphically using lasso tool (don't forget to set selection level to residue).
This selection is used solely to define boundaries of the docking search and the size of the
grids and doesn't have to be complete, selecting some 4 residues delimiting the binding site
is sufficient. Receptor setup dialog also lets you run binding site identification routine to
quickly locate putative binding sites on your receptor.
The receptor setup procedure will first display the grid box, allowing you to adjust the box dimensions, and
then the 'probe' which defines the initial positioning of the ligand's center of mass and
long/short axis. The probe can be moved/rotated. While its positioning has only minor
influence on the results as long as it remains inside the binding site, it may help the
procedure to find the correct docked orientation more reliably and/or in shorter time.
After the receptor setup is complete, the program normally displays the receptor with the
selected binding site residues highlighted in yellow xstick representation.
Ligand setup offers a number of ways (submenu docking/ligand setup ) to define the ligand,
depending on the source of the ligand structure(s).
To run the docking job from a unix shell, use the _dockScan macro with appropriate parameters, e.g.
$ICMHOME/icm -c /icm/_dockScan abl -a -S confs=10 effort=2. from=10 to=20 outdir=/tmp/ name=ou >& abl10.ou &
The _dockBatch script streamlines the setup and execution of docking simulations, enabling automated preparation of the docking project and optional immediate launch of screening runs. This tool supports flexible definition of the binding site, ligand treatment, and docking mode through a range of command-line arguments:
$ICMHOME/icm $ICMHOME/_dockBatch <receptor.pdb|receptor.ob> [ligands2dock.sdf] [proj=projectName] <options>e.g.
$ICMHOME/icm $ICMHOME/_dockBatch 1xbb.pdb ligmol=astiArguments:
- ligmol=
: ligand in the input structure, used to define site (or redock with -r) - pocket=
: pocket selection in a_chain/resNumList format, e.g. a_b/5,8:10,115 - pocket=
: pocket selection will be defined by a 3D ligand from an .sdf file - box=x,y,z,X,Y,Z : site definition as a box, e.g. -1.,3,-10.,10.,13.,0. Our box definition is x,y,z,X,Y,Z which is to say two corners of the box (with lowest/HIGHEST xyz coordinates). If you want to use center Xc,Yc,Zc and size (box dimension) d you could calculate xyzXYZ as Xc - d/2 Yc - d/2 Zc - d/2 Xc + d/2 Yc + d/2 Zc + d/2
- -r : re-dock mode, takes ligand from the receptor complex input
- -R : same, with RMSD calculation
- -u : do not automatically set protonation/charge state of the ligand
- -w : delete all water in the receptor input
- -e : use stack embedded in receptor as ensemble for '4D' docking
- recConf=
: use specific receptor conformation - -dnaChargeScale : scales down charges for phosphate group
- covres=
: covalent docking mode, modified residue in a_chain/resNum format - react=
: table with custom covalent reaction mechanisms - reactNum=
: selects reaction from multiple mechanisms in the default or custom table - template=
: icm object file with ligand template to be used to bias docking - tmplMeth=
- proc=N : run multiple ligands across N threads/CPU cores in parallel
- pocket=
Fully Automated Optional - you can run screening automatically by defining the sdf file at the end
e.g.
$ICMHOME/icm64 $ICMHOME/_dockBatch 1xbb.pdb ligmol=asti chem_database.sdfNote about Ionization State of Ligand setup using _dockBatch There is a setting within the docking project that controls protonation state handling. When project is set up by _dockBatch , the default is to apply built-in pKa model to determine predominant protonation state. To keep protonation as in input sdf, you can turn this off with _dockBatch -u option (or change it in .dtb file later). If you just want to see results of automatic protonation state assignment, you can load your sdf in icm and use dialog under Chemistry/Set Formal Charges
convert command to
a pdb-entry with ligands, the ligands will just become some crippled incomplete molecules
which can not be further conformationally optimized.
Follow these steps to convert a chemical properly from a pdb form to
an a correct icm object.
- display the molecule, set
wireStyle=2(or via top-left gui-menu), and selection type toGRAPHICS.selectionMode=1(the first item of the gui-selection-mode menu) - invoke
MolMechanics.Structure.SetBondTypemenu item - graphically select groups of atoms (e.g. a ring) and set appropriate bond type
- invoke the next menu item,
MolMechanics.Structure.SetFormalChargeand set formal charges - proceed to the
MolMechanics.ICM-Convert.Chemicalmenu (see below)
Setting up a ligand or a set of ligands
Let's now consider the situation when icm object of the ligand loaded. ICM object of the ligand can also be prepared, for instance, by reading structure from SD file (menu
File/Read Molecule/Mol/SDF ) and converting it to ICM
(menu MolMechanics/ICM-Convert/Chemical ).
Once the icm object of the ligand is ready, proceed to docking ligand setup (menu
Docking/Ligand Setup/From Loaded ICM object ).
Ligand setup procedure can be ran repeatedly to change the ligand source within the same docking project. Also box size and probe position can be changed later (menu
Review/Adjust ligand/box ).
At this point, the project is ready for the calculation of maps (menu docking/Make receptor maps ).
The recalculation of the maps typically requires less than 1 minute.
While the map calculation dialog allows changing the grid step, we do not recommend altering
the default value of 0.5 which was found optimal for a large number of test cases.
Maximum van der Walls repulsion parameter can be increased if more rigorous enforcement of steric exclusion is desired
With the map calculations completed, everything is ready to start the actual docking
simulation.
A larger set of ligands in a mol file can be considered as a database and indexed with the ICM indexing tool
(menu Docking/Tools/Index Mol,Mol2 Database ) for fast access. Ligand structures from mol/mol2 file
can be converted to ICM on the fly and do not require manual preparations necessary in the case of PDB structures.
docking/Small Set Docking Batch to start docking of one or few ligands in the
background. You can also view the process interactively (menu docking/Interactive Docking )
although it is much slower due to the time spent on drawing the molecules. The results of the
batch docking job are saved in the
PROJECTNAME_answers*.ob #icm-object file with best solutions for each ligand
PROJECTNAME_*.cnf # icm conformational stack files with multiple docked conf.
PROJECTNAME_*.ou # output file were various messages are stored.Docking/Browse Stack Conformations ). Use menu
Docking/Display/Preferences to change default graphic representation of ligand/receptor.
Docking/Tools/Index Mol/Mol2 File/Database to generate the index, then set up the SDF/MOL2
file as a ligand source (menu Docking/Ligand Setup/From Database ). As in docking, _dockScan
ICM script can be ran directly from UNIX shell/command line to start simulations.
#>r DOCK1.r_ScoreThreshold
-35.The choice of the threshold can be done in two ways:
- based on the scores calculated by docking known ligands. Generally, a value somewhat above typical score observed for known ligands is a good guess.
- if no ligands are known, a pre-simulation can be run using ~1000 compounds from the target database. Using the resulting statistics for the scores, the threshold should be set to retain ~1% of the ligands.
icm.pmf file
and read with the read pmf s_pmfFile command.
There are two types of the mf-calculation: all-to-all atoms and intermolecular mode.
The mode is switched with the mfMethod preference.
To enable calculation of the pmf-score, define the PROJECTNAME.r_mfScoreThreshold
threshold paramter to the table:
#>r PROJECTNAME.r_mfScoreThreshold
999.
#>i DOCK1.i_maxHdonors
5
#>i DOCK1.i_maxLigSize
500
#>i DOCK1.i_maxNO
10
#>i DOCK1.i_maxTorsion
10
#>i DOCK1.i_minLigSize
100icm _dockScan from=1 to=10000 MYPROJECT
icm _dockScan from=10001 to=20000 MYPROJECT
icm _dockScan from=20001 to=30000 MYPROJECT
..qsub $ICMHOME/pbsrun -v"JOBARGS=-f 1 -t 1000 -o MYPROJECT"
Note that the rundock arguments go in the quotes after JOBARGS= . The qsub command is a part of PBS.
To submit multiple jobs, there is a simple shell script 'pbsscan' which executes multiple qsub's for database stripes:
$ICMHOME/pbsscan MYPROJECT 1 6000 1000
-submits 6 jobs, 1 to 1000; 1001 to 2000 ... 5001 to 6000. Currently this script only supports default rundock arguments, copy/edit to change.
The command qstat is a part of PBS and can be used to check the status of the jobs. In addition, $ICMHOME/scanstat script can be used to monitor the progress of the VLS jobs. It analyses the *.ou rundock output files.
$ICMHOME/scanstat *.ou
To delete the jobs, use PBS command qdel:
qdel 1234 # delets job number 1234
The MFScore is calculated if
r_mfScoreThreshold
variable is defined in the project .dtb file. It can be added manually:
#>r PROJECTNAME.r_mfScoreThreshold
999.
The hitlist can also be prepared by a macro. In this case the scores will be extracted.
Docking/Make Hit List... ).
An older way to export hits as SD file is using
(menu Docking/Tools/Export scan answers as mol ).
The score and its components are stored in the resulting SD file as well.
Simple analysis of the score distribution can be performed by making a histogram
(menu Docking/Tools/Scan results histogram ).
To make a hitlist in GUI use Docking/Make hitlist and on the command line use _scanMakeHitlist
scanMakeHitList "DOCK1" ""//vls/DOCK1_answers*.ob" Name(Name( "//vls/DOCK1_answers*.ob"")) no no yes 0The logical arguments at the end are:
- l_import2DfromDB (no) - you can make the hitlist smaller by not saving 2D
- l_makeUnique (no) - if you had made mulitple runs you can choose the highest scoring ligand
- l_import3D (yes) - will import 3D coordinates
- i_topScored (0) - if you had a large hitlist you can choose to import just the top hits e.g. top 1000 based on score.
Please see the GUI Manual for a description of the physics-based score (Score) and the Neural Network score (RTCCN).
After the project, the project directory and the maps have been created, you can start docking different sets of ligands into this receptor. To run it directly by ICM instead of through an intermediate Unix shell script, use the _dockScan script.
To run the _dockScan script just run ICM and provide the script as the first argument. All _dockScan arguments need to be provided after it.
Prerequisites:Complete these steps of the Docking menu:
- Receptor Setup
- Make receptor maps
- Ligand setup ( Note: you do not need this step if you dock directly from an .sdf file)
DOCK1.dtb) to your liking if necessary.
The full syntax of the _dockScan script is the following.
icm _dockScan [ optional arguments ] projNameThe arguments could be the following
| argument | comment | example |
|---|---|---|
| dock according to parameters and ligand source settings from the file projName.dtb | ||
input= | dock directly from an sdf file (other modes require input specification in the PROJECT.dtb file)
| |
input= | dock peptides from an se file containing sequence(s) of aminoacid residues, which may have modifications (modres)
| |
-a | dock and save ALL molecules, ignore filters and score threshold (default if less than 100 molecules) | icm $ICMHOME/_dockScan -a /home/dock/PROJECT |
-d | docking only, NO scoring | |
-E | evaluates binding score for several poses, resorts them but does save all the stacks in files (convenient for screening) -S | |
-p | "probe" mode: dock probes from the input sdf file, encourage spacial coverage | |
-r | RIGID ligand docking | |
-s | save stacks of multiple docking poses for each ligand | |
-S | same as -s but reSCORE ALL saved poses, see also -E | |
-f | apply property filters to exclude molecules outside ranges specified in PROJECT.dtb (this is the default if there are more than 100 molecules being docked) | |
confs= | score/save only up to | |
-n | generate and score near-native conformation as provided in input | |
-Cn | explicit option to auto assign formal charges according to NN pKa model | |
from= to= | range of molecules from an indexed .sdf file or molcart ids | from=1 to=10000 |
jobs= | spaws n processes, default is 1 | |
name= | the result file with poses will be named accordingly (the default is 'answers') | |
outdir= | directory for the output files | |
output= | process answers.ob into a hitlist sdf file at the end of the run | |
seed= | random seed from the previous docking to reproduce the results exactly | |
effort= | a 'thoroughness' factor that allows one to extend the docking effort or reduce it (default is 1., reasonable range from 0.5 to 20. ) | |
proc= | run specified number of parallel jobs (eg use #cores ) | |
scoreCutoff= | accept ligs with score better than |
Example:
Docking an sdf file (first configure the receptor, make the grid maps and setup the ligand input source in GUI).
icm _dockScan /home/gpcr/PROJECTNAME -a -S confs=10 effort=3.This will dock all compounds with 3-fold longer (more thorough) simulations, and rescore up to 10 conformations per ligand.
If you have a cluster license without graphics you will need to use -vlscluster flag after calling icm.
icm -vlslcuster _dockScan /home/gpcr/PROJECTNAME -a -S confs=10 effort=3.
l_superByName controls the way
correspondence is established between the ligand and template atoms. If it is
'no', chemical substructure search is performed and tethers imposed according to
the substructure match. If l_superByName is 'yes', simple matching according to
atom names is performed. Tethers can be individually weighted by assigning
b-factor values to the template atoms. Weights are reversely proportional to b-factor,
default b-factor of 20. corresponds to the weight of 1.
In the /bin directory you will find a script called docksub.icm. This script prepares your docking run and distributes it via SGE or SLURM job queueing system on your cluster or cloud.
use:
$ICMHOME/icm docksub.icm -vlscluster <chemTable.sdf|.inx> {<dockProj>|<APF_template.mol>} [jobs=100] [-sub] [-apf] [qtype=sge|slurm] [<dockProj_and_dockScan_options>|<APF_options>]e.g. submit 100 slurm jobs
icm64 docksub.icm qtype=slurm chemTable.inx DOCK1 jobs=100 –subicm64 docksub.icm input.inx DOCK1 jobs=2 proc=18
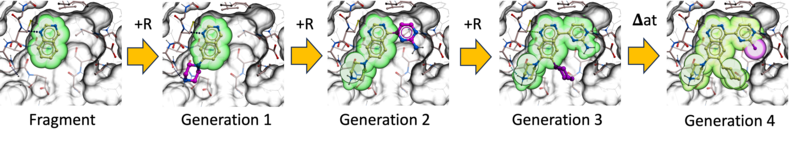
Ligand AIDE is a de novo ligand generation workflow based on Artificial Intelligent Design Evolution. It uses an iterative evolutionary strategy to grow and optimize ligands directly in the context of a protein binding site.
The process begins with a population of docked small fragments. From this starting point, new designs are generated by adding or replacing R-groups using the groupGen neural network, as well as by performing atom-level substitutions using the Atom Predictor. Each newly generated cohort of compounds is then re-docked into the binding site.
Designs are refined through multiple evolutionary cycles, typically three to five iterations. At each step, a Darwinian selection process removes poor designs based on several criteria, including RTCNN scores, physics-based binding scores with penalty terms, drug-like property filters, and synthesizability.
Through successive generations, fragments evolve into more complete and optimized ligands. This iterative grow-and-select strategy enables efficient exploration of chemical space while maintaining binding quality, drug-like behavior, and practical synthetic feasibility.
To run:
- Setup the docking project.
- Use the dndAI.icm script (see below) in $ICMHOME/bin
Usage> icm64 dndAI.icm prj=
GINGER - Graph Internal-coordinate Neural-network conformer Generator with Energy Refinement
Read More...
Usage> icm64 _ginger input.sdf|.tsv|.csv header=no smicol=A idcol=B output.sdf|.molt
Example:
RIDGE (Rapid Docking GPU Engine) is an extremely fast and accurate structure-based virtual ligand screening method.
RIDGE combines MolSoft's experience and knowledge in GPU programming to create a docking engine that fully runs on the GPU. The central part of the engine is fully RAM and GPU optimized, and it also utilizes low-level CPU multithreading to ensure a balanced workload between the CPU and GPU. Additionally, it is fully compatible with existing ICM docking projects, requiring no extra setup.
Read More...
Options:
We recommend to use if possible a most recent NVIDIA GPU. For optimal performance you would use a GEFORCE 4090 but RIDGE will run on older versions (e.g. 3090 2080).
Please note multiple .molt files can be screened using comma separated filenames.
Example
Dock 1000 random compounds from /path/to/conf_db.molt with Cartesian minimization (-C)
CombiRIDGE is a new innovative GPU-accelerated solution for high-throughput ligand docking and screening which leverages MolSoft's generative neural network conformer enumeration method GINGER, ultra-fast GPU docking technology RIDGE and advanced graph neural network scoring RTCNN. The approach allows you to optimize specific R-groups and screen vast ultra-large or combinatorial libraries efficiently.
Options:
Unpack the files:
Create a link to MEL library and Markush
Create your docking Project
You can setup your docking project files in the GUI or fully automated in the command line using _dockBatch
STEP 1 - Minimal Enumeration Library (MEL) Docking
Copy docking project files to the 'run' directory
Submit docking job(s) in 'run' directory
Single Machine
Cluster using docksub.icm script in /bin directory
At the end of this stage you expect multiple answer files in the 'run' directory.
STEP 2.1 (Load and Process Hits)
STEP 2.2 (Enumerate)
STEP 3 (Dock final hits)
The GigaScreen method combines machine learning and deep learning tools to tackle the computational intensity of screening very large chemical databases. To overcome these challenges several protocols are employed:
Read more here about GigaScreen.
System Requirements
How to run GigaScreen:
1) Download and install icm-mxnet package - contact MolSoft to download this package and a RIDGE GPU license is required.
Make sure you can run the binary (no missing dependencies)
1.1) Download compressed conformation DB - contact MolSoft to obtain these databases.
2) gigaScreen.icm script is provided with distribution ($ICMHOME/bin/gigaScreen.icm)
2.1) create working directory and copy docking project files
# docking project consists of following files (copy them into working directory)
2.2) run gigaScreen.icm inside working directory
That should perform 5 iterations of RIDGE/Build Model/Predict + Final Docking
All intermediate results will be stored inside 'screen_out' directory
Each stage will create the following files:
* ridge_out_
Final Docking will be saved as screen_out/ridge_final.sdf
Overview:
Conformer generation is an essential step of a variety of molecular modeling and computer-assisted drug discovery workflows such as 3D ligand-based virtual screening or fast GPU docking. GINGER (Graph Internal-coordinate Neural-network conformer Generator with Energy Refinement) is Molsoft's new cutting-edge software designed for ultra-rapid high quality conformer library generation on GPUs.
icm64 _ginger input.csv outut.sdf header=yes -f smicol=mol idcol=id sdfcompress=yesRIDGE - Rapid Docking GPU Engine
$ICMHOME/icm $ICMHOME/_ridge DockingProjectName <projFile> output= .sdf [<options>] input= DB_3D_Confs.molt
$ICMHOME/icm64 $ICMHOME/_ridge <mydockProj> -C input=/path/to/conf_db.molt randomSelect=1000icm <dockProj> [input=<MEL.sdf] [space=<molsoft_space.icb>] [output=<.sdf|.icb] [fr=<i_fragFrom>] [to=<i_fragTo>] [mnhits=<N_tophits>] [mnmol=<N_Enumeration_Limit>] [effort=1.] [-S] [-C]
cd vsyn_example
ln -s /path/to/files_MEL_example files # or files_MEL_all
ln -s /path/to/markush markush
Create directory 'run' in the main 'vsyn_example'
mkdir run$ICMHOME/icm64 _dockScan <dockproj> input=../files/files_MEL_275592_.inx effort=2. scoreCutoff=-18. name=files_MEL_275592_ proc=<n_parallel>
<c>
$ICMHOME/icm64 _dockScan <dockproj> input=../files/files_MEL_2comp_.inx effort=2. scoreCutoff=-18. name=files_MEL_2comp_ proc=<n_parallel>$ICMHOME/icm64 docksub.icm ../files/files_MEL_275592_.inx <dockproj> jobs=<n_jobs> effort=2. scoreCutoff=-18. name=files_MEL_275592_ proc=<n_proc>$ICMHOME/icm64 docksub.icm ../files/files_MEL_2comp_.inx <dockproj> jobs=<n_jobs> effort=2. scoreCutoff=-18. name=files_MEL_2comp_ proc=<n_proc>cd .. # go to main 'vsyn_example' directory)
$ICMHOME/icm64 icm_load_hits_enamine_CARC_062021.icm
bash CapSelectMP_full.sh# Default number of output molecules is 2500000
# This can be changed by editing script icm_enumerate_REAL_frags_112021_chunkify_charge_CARC.icm
# MAX_MOLS="2500000"
$ICMHOME/icm64 icm_enumerate_REAL_frags_112021_chunkify_charge_CARC.icm
# Result files will be placed into
run/processing_files/enumerated_best_frags_<dockproj>__<MAX_MOLS>/*.sdf
cat run/processing_files/enumerated_best_frags_<dockproj>__<MAX_MOLS>/*.sdf > run/enumerated_best_frags_all.sdf
sh icm-mxnet-3.9-3b-linux.sh -p/path/to/icm393b
export ICMHOME=/path/to/icm393b$ICMHOME/icm64mkdir giga_test
cd giga_test$ICMHOME/icm64 gigaScreen.icm project=<dock_proj> niter=5 /path/to/REAL/*.molt