[ Graphics | Simulations | Sequence | Modules ]
Let us go through the short overview of the ICM application areas.[ Views | Skin | RNA/DNA | Combos ]
ICM and ICM-derived plugins provide a viewing environment for large a small molecules and
general three-dimensional objects with or without textures. Various types of enhancements including stereo, anti-aliasing, graphical layers, on-the-fly generation of shadows, occlusion shading, custom backgrounds, depth cueing and simple rotation, translation, zooming, clipping, picking, continuous movements, separate
The views include
|

|
| The contour-buildup algorithm calculates the smooth and accurate analytical molecular surface in seconds. This surface can be saved as a geometrical object, saved as a vectorized postscript file. |

|
The skin is used in the REBEL algorithm to solve the Poisson equation, as well as in the molecular surface analysis routines (e.g. a projection of physical properties on the receptor surface ).
Also ICM can build and draw a solvent-accessible surface ( see surface ) and
* a Gaussian molecular density which can be contoured
at different levels and to generate different smooth molecular envelopes
and enclosed pockets and cavities:
|

|
PDB entry: 101d
ICM command: |
|
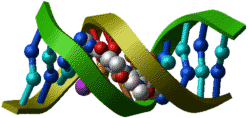
PDB entry: 4tna
ICM commands: |

|

|
Simplified molecular representations are built automatically (e.g. the protein-dna complex is shown with one command: nice "1dnk" ).
You can combine different types of molecular representations
with solid or wire geometrical objects.
|
[ Peptides | Homology | Design | Symmetry | Protein-Protein Docking | Pockets | Docking & VLS | 3D chemical editing | Electrostatics ]

| Take a peptide sequence and predict its three-dimensional structure. Of course, success is not guaranteed, especially if the peptide is longer than about 25 residues but some preliminary tests are encouraging. |
_folding file and go ahead.

|
ICM has an excellent record in building accurate models by homology. The procedure will build the framework, shake up the side-chains and loops by global energy optimization. You can also color the model by local reliability to identify the potentially wrong parts of the model. |
ICM also offers a fast and completely automated method to build a model by homology and extract the best fitting loops from a database of all known loops (see build model and montecarlo fast). It just takes a few seconds to build a complete model by homology with loops.

|
ICM was used to design two new 7 residue loops and in both cases the designs were successful. Moreover, the predicted conformations turned out to be exactly right (accuracy of 0.5A) after the crystallographic structures of the designed proteins were determined in Rik Wierenga's lab. Use the _loop script to predict loop conformations and
calcEnergyStrain to identify the strained parts of the design.
|
ICM has a database of loops compiled from PDB structures. To search a fragment against the database and load the conformers into a session (eg into a stack), use the following command:
build loop stack rs_fragment .
The display stack command will allow to view the conformers. You may also load the stack into the object with the
load stack a_ command.

|
ICM has a full set of commands and functions to generate symmetry related molecules and generate "biological units". |
|
As demonstrated in several recent papers, short flexible peptides can be successfully docked ab initio to their receptors. This method is a blend of the peptide folding with the grid potentials representing the receptor. A similar method can be applied to any chemical. A chemical can be built from a 2D representation and optimized. The "druggable" pockets can be predicted with an algorithm based on the contiguous grid energy densities. |

|
In virtual screening the flexible docking is applied to hundreds of thousands of individual ligands. This version of docking is fast and requires an accurate relative binding or ranking function to discriminate between the true ligands and hundreds of thousands of potential false positives. The ligand sampling and docking procedure is a combination of the genuine internal coordinate docking methodology with a sophisticated global optimization scheme.
| Accurate and fast potentials and empirically adjusted scoring functions have led to an efficient virtual screening methodology in which ligands are fully and continuously flexible. |

|
ICM allows one to draw a molecule directly in 3D with full undo/redo support and check its fit to a protein binding pocket. This environment is called a 3D ligand editor. The editor functionality is described in the User Manual.
ICM incorporates a very fast and accurate boundary element solution of the Poisson equation to find the electrostatic free energy of a molecule in solution. This algorithm (abbreviated as REBEL ) can be used dynamically during conformational search. The components of the electrostatic free energy are used to calculate the binding energy and evaluate the transfer energy between water and organic solvents.
ICM uses generalized Born approximation to calculate the electrostatic solvation energy and its gradient dynamically during local and global conformational searches.
| The electrostatic potential can be projected on a molecular surface for the identification of possible binding sites. |

|
[ Genomics | Dotplot | Alignment | Multi-alignment | Trees,clusters,evolution | Searches | Plotting ]

Handling gigabytes of genomic sequence, fast cross-comparison of millions of sequences was another challenge solved in the ICM program. ICM can identify a unique subset of millions of sequences, assemble sequences from Unigene clusters into alignments ( SIM4 program is used a part of the procedure).
Using the plotSeqDotMatrix macro:
read sequence s_icmhome + "zincFing.seq"
plotSeqDotMatrix 2drp_d 3znf_m \
"Two z-finger peptide" "Human Enhancer Domain" 5 20
(if the macro complains about s_psViewer , set it in Preferences/Directories , or reassign, e.g. s_psViewer = "display" )
Here the color shows the local significance of the alignment. You can change the method to calculate probability, color scheme and residue comparison matrices and calculate it interactively or in batch. |

|
alignment). The ZEGA probability is a more sensitive
indicator of structural significance than the BLAST P-value.
The structural statistics was derived by
Abagyan and Batalov, 1997:
read sequence s_icmhome + "sh3.seq"
show Align(Fyn Spec) # the probability will be shown
The ICM alignment functions and commands are summarized in the alignment section.
Read any number of sequences in fasta or swissprot formats and automatically align the sequences, interactively or in a batch. It will look like this:
# Consensus ...#.^.YD%..+~..-#~# K~-.#~##.~~..~WW.#. ~~.~G%#P.
Fyn ----VTLFVALYDYEARTEDDLSFHKGEKFQILNSSEGDWWEARSLTTGETGYIPS
Spec DETGKELVLALYDYQEKSPREVTMKKGDILTLLNSTNKDWWKVE--VNDRQGFVP-
Eps8 KTQPKKYAKSKYDFVARNSSELSM-KDDVLELILDDRRQWWKVR---NSGDGFVPN
# nID 7 Lmin 56 ID 11.5 %
#MATGAP gonnet 2.4 0.15read sequence s_icmhome + "sh3.seq"
group sequences sh3
align sh3
show sh3The gui version of ICM also has a multiple alignment viewer with dynamic coloring according to conservation tables CONSENSUS and CONSENSUSCOLOR. It will automatically show secondary structure and other features.

The ICM alignment functions and commands are summarized in the alignment section.
The Algorithm
The multiple sequence alignment is produced using hierarchical clustering of the sequences based on sequence similarity calculated with the ZEGA alignment (a modification of the Needleman and Wunsch algorithm permitting zero gap-end penalties, ZEGA alignment) and Gonnet et al., residue substitution matrix [gon92]. The branches are then aligned in the tree order to form profiles that are progressively aligned to other profiles or sequences to form multiple sequence alignment.
Relationships between sequences can be presented in three
forms:
|

|
ZEGA alignment (full dynamic programming with zero end gaps) is used
for each comparison and an empirical probability function described in JMB,1997 is used to assign a P-value to each hit.
This search may give you more homologs that a BLAST search!
The output may presented in a linked table form:
| NA1 | NA2 | ID | SC | pP | DE |
| Fyn | 1nyf_mNo | 100. | 62.81 | 20.94 | fyn |
| ... | lines skipped | ... | ... | ... | ... |
| Eps8 | 1tud_m17 | 21. | 17.04 | 4.17 | alpha-spectrin |
| Eps8 | 1fyn_a23 | 22.6 | 17.02 | 4.16 | phosphotransferase fyn |
| Eps8 | 1efn_a25 | 22. | 16.64 | 4.11 | fyn tyrosine kinase |
| Eps8 | 1hsq_mNo | 24.2 | 16.87 | 4.1 | phospholipase c-gamma (sh3 domain) |
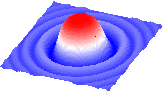
Take a matrix and represent it in 3D in a variety of forms.
View it in stereo, color, label, transform with the mouse.
Example:
|
|

|
- ICM-browser and ICM-browser-pro (distributed from a single package)
- ICM-chemist and ICM-chemist-pro (distributed from a single package)
- ICM-pro with options including bioinformatics, Poisson electrostatics, chemistry and cheminformatics, homology modeling, docking, virtual ligand screening (VLS).
- Windows
- Linux and Unix
- Macintosh
The modules have the following features:
ICM-main
- shell for molecules, numbers, strings, vectors, matrices, tables, sequences, alignments, profiles, 3D maps, 3D graphical objects, 2D chemical tables/spreadsheets, images
- ICM-language and macros
- graphics, stereo
- imaging and vectorized postscript
- animation and movies
- mathematics, statistics, plotting
- presentation of the results in html format
- user-defined and automated interpretation of web links
- HTML-form-output interpretation
- pairwise and multiple sequence alignments, evolutionary trees, clustering
- secondary structure prediction and assignment, property profiles, pattern searching
- superpositions, structural alignment, Ramachandran plots
- protein quality check
- analytical molecular surface
- calculations of surface areas and volumes
- cavity analysis
- symmetry operations, access to 230 space groups
- database fragment search
- identification of common substructures in PDB
- read pdb, mol2, csd, build from sequence
- energy, solvation, MIMEL, side-chain entropies, soft van der Waals, tethers, distance and angular restraints
- local minimization
- ab initio peptide structure prediction by the Biased Probability Monte Carlo method
- loop simulations
- side-chain placement
ICM-REBEL (electrostatics)
- electrostatic free energy calculated by the boundary element method
- coloring molecular surface by electrostatic potential
- binding energy (electrostatic solvation component)
- maps of electrostatic potential and its isopotential contours
ICM-docking
- indexing of chemical databases in SD, mol2 and csd format
- searching and extracting from the indexed databases
- fast grid potentials
- scripts for flexible ligand docking
- scripts for protein-protein docking
- 2D (SMILES) to 3D conversion, type and charge assignment, mmff geometry optimization, low-energy rotamer generation
- refinement in full atom representation
ICM chemistry (also, see here)
- scripting access to the internal chemical spreadsheets and external chemical databases in MySQL (via
molcart) and SQL lite. - Various operations on chemical tables (see below)
- integration with the docking engine and interactive ligand editing
Operations on chemical tables:
- Calculate various properties and descriptors from 2D chemical table
- Standardize chemical structure (change ambiguous depictions of some functional groups to a standard form according to user defined tables)
- Build QSAR type prediction models from a chemical spreadsheet and apply those models to new chemical tables
- Convert Smiles to 2D and the opposite operation
- Generate 2D Depiction from a 3D or 0D chemical
- Convert a 2D chemical to 3D
- Generate 3D Conformers
- Generate Tautomers
- Generate Stereoisomers, assign and manipulate stereo centers
- Align/Color By 2D Scaffold
- Cluster chemicals by either fingerprint similarity or external distance matrices
- Compare two chemical sets for common elements
- Sort a table and select duplicate rows in a table
- Create/Modify Markush objects
- Enumerate a combinatorial chemical library from scaffold and R-groups
- R-Group Decomposition of a chemical spreadsheet
- Enumerate a chemical library by reaction(s) and reactants
- Various forms of multiple chemical superposition (both 2D and 3D)
ICM-bioinformatics
- fast comparison and redundancy removal of millions of genomic or protein sequences
- multiple EST clustering, alignment and consensus derivation
- database indexing and manipulations
- functions to evaluate sequence-structure similarity
- scripts to recognize remote similarities in the protein sequence and PDB databases
- search a pattern through a database
- searching profiles and patterns from the Prosite database through a sequence
- HTML representation of the search results with interpretation of links
- interactive editor of sequence-structure alignment
- automated building of models by homology with loop sampling and side-chain placement (fast homology model building combined with the database loop search is a separate module which is ICM Homology).
ICM-Homology
- sequence-structure alignment (threading)
- ultra-fast automated homology model building with a database loop search
- loop modeling and refinement, side-chain placement
- surface analysis
Abagyan and Totrov, 1994),
pseudo-Brownian docking method ( Abagyan, Totrov and Kuznetsov, 1994)
and local deformation loop movements ( Abagyan and Mazur, 1989 ).
A set of ECEPP/3 energy terms is complemented with the parameters for rare atoms and atom types, as well as the solvation energy terms, electrostatic polarization energy and side-chain entropic effects (
Abagyan and Totrov, 1994),
making the total calculated energy a more realistic approximation
of the true free energy.
The MMFF94 force field has also been implemented.
Powerful molecular graphics, the ICM-command language,
and a set of structure manipulation tools and penalty functions (such as
multidimensional variable restraints, tethers, distance restraints) allow
the user to address a wide variety of problems concerning biomolecular
structures.