| Video |
Refining a virtual screening hitlist with molecular dynamics (MD) simulations can be highly beneficial, particularly when assessing the stability and realistic binding behavior of candidate compounds in complex with the target. While initial docking provides a static snapshot of ligand-protein interactions, MD allows for the exploration of dynamic, time-dependent effects, such as conformational flexibility and solvent effects, which are crucial for accurate binding predictions. Through MD, we can observe how ligands adjust within the binding pocket, monitor changes in the protein's structure, and detect transient interactions that might be missed in docking alone. By refining the hitlist with MD, we can prioritize compounds that not only bind well in static models but also exhibit sustained interactions in a more realistic, fluctuating environment. This extra layer of validation helps improve the reliability of hitlist rankings and reduce the risk of advancing false positives in drug discovery.
To run use the script runMDopenMM which is in /bin directory of the ICM distribution use the commands below in the terminal window. In preparation you need to save your hitlist in .icb format (right click on the hitlist table tab and choose Save as... .icb) and you need to save your receptor as an ICM object.
$ICMHOME/icm64 $ICMHOME/bin/runMDopenMM.icm -w time=5.0 confs=20 -m -rl ligfile=<hitlist>.icb <receptor>.ob
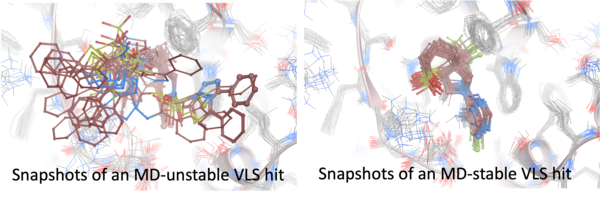
Ligands are scored using the ICM physics-based scoring method and the RTCNN neural network score, with RTCNN offering faster performance. Evaluation primarily focuses on the stability of the pose and its RTCNN score. The MD_RMSD0_mean metric is used to measure deviations from the initial pose, with preferred ligands remaining within approximately 1.0-1.2 Å. This threshold may be adjusted based on the binding site, being more stringent for enclosed pockets and more flexible for open sites. Stable complexes are characterized by maintaining favorable interactions and consistent scores during the simulation, which can be assessed using the MD_RTCNN_mean metric.
Guide to the results column:
- RMSD0 is versus initial pose supplied (from docking), i.e. how much it moves away from docking pose.
- RMSD is versus the snapshot after 1/3 of the simulation time. The idea is that if the pose gets indeed ‘refined’ it should stabilize reasonably soon.
- _tcov values are time covariances, i.e. indication of continuous drift towards higher (or lower) values.
- Success When running large numbers of runs there might be a small fraction of runs that end in some kind of numeric failures, with no trajectory produced. These are marked with 0 in ’success’ column.