Google Search the Manual:
Keyword Search:
| Prev | ICM User's Guide 12.11 ICM X-Ray AutoFit - Automated Model Building into Density | Next |
| Available in the following product(s): ICM-Pro | ICM-VLS | |
The ICM X-Ray AutoFit is an automated method to fit a ligand into electron density. The tool combines the powerful ICM docking algorithm with an electron density fitting function.
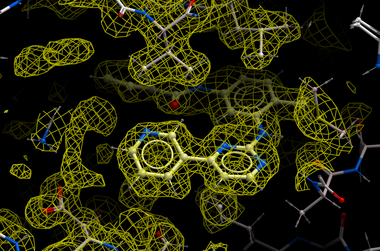
The input for ICM X-Ray AutoFit is an electron density map in CCP4 format, the protein recpeptor and ligand which can either be drawn or imported into ICM.
Theory
The ICM X-Ray AutoFit method includes the following features:
- Soft docking energy function: Traditional rigid docking methods may struggle when the binding pocket exhibits some flexibility. The soft docking energy function accounts for minor conformational changes, allowing a better fit within the pocket and avoiding artificially high strain on the ligand.
- Intra and inter ligand interaction energy function: The method considers both the internal strain of the ligand and how it interacts with surrounding residues in the binding site. This ensures that the final pose not only fits the density but is also chemically reasonable and energetically favorable.
- Weighted electron density contributions: Not all parts of the electron density contribute equally to the ligand placement. This method applies a weighting scheme to prioritize high-confidence regions of the density, improving the accuracy of the fit.
- Electron density filtering to exclude protein receptor atoms: To prevent interference from the protein structure itself, the electron density used for ligand fitting is filtered. This step removes areas occupied by receptor atoms, ensuring that only relevant density is considered for ligand placement.
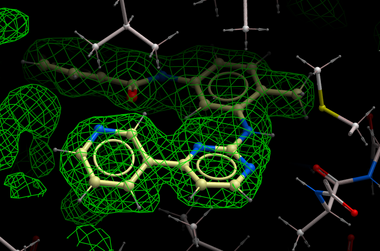
| NOTE: Density from the receptor is automatically filtered out from the analysis. In the picture shown above the green map represents attractive potential. |
The method generates multiple hits for each ligand with a score assigned. It has been demonstrated that improved ligand receptor interactions can be determined by the ICM X-Ray AutoFit method compared to published crystal structures.
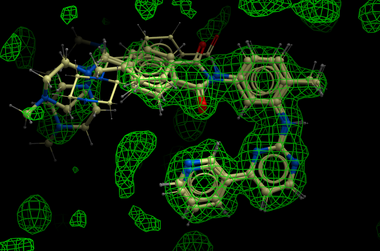
In the figure shown below the interaction between Gleevec and Syk kinase is shown. The white carbon atoms are the published ligand pose and the yellow carbon ligand is the result of ICM which gives a better fit to the density.
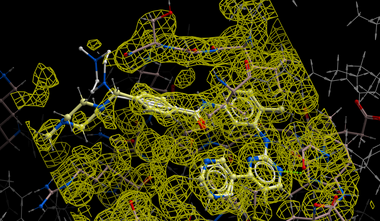
Instructions
How to run the ICM X-Ray AutoFit.
- Load the receptor structure and convert to an ICM object.
- Load the CCP4 map (File/Open) or download the map from the PDB using Tools/X-Ray/Get Electron Density Map.
- Load the ligand and convert to an ICM object.
- Follow the small molecule docking procedure but after generating maps select Docking/X-Ray Density and then undertake docking in the standard way. When an X-Ray Density is selected the field in the DockProj.dtb r_densityWeight will be set to 1. You will also see an additional map of the electron density in your docking directory called DockProj_xr.map.
- Dock the ligands in the regular way either from .sdf or chemical table.
| Prev AI Generator | Home Up | Next Peptide Docking |