Google Search the Manual:
Keyword Search:
| Prev | ICM User's Guide 10.5 Chemical Search | Next |
[ Query | Filter Search | Processing | Text | Giga Search | Pharmacophore Search ]
| Available in the following product(s): ICM-Chemist | ICM-Chemist-Pro | ICM-VLS |
Chemical similarity searching can be used to screen a database of compounds for structural similarity to a query chemical structure. The chemical similarity search window is shown below.
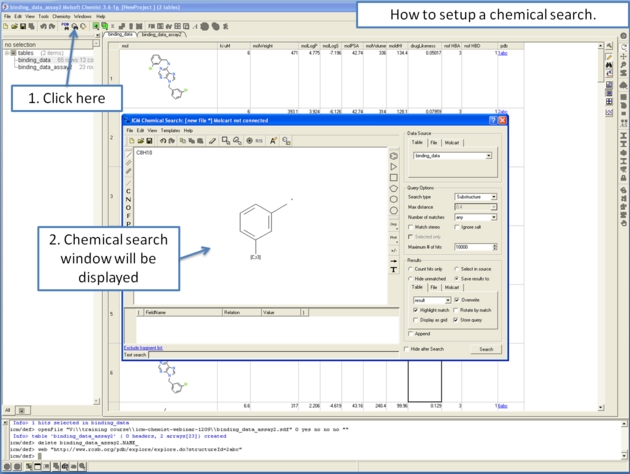
To access this window use the Tools/Chemical Search menu or select the button shown below.
10.5.1 Query Setup |
To set up a query first you must have either drawn or loaded a chemical structure into ICM. Instructions for this are described in the Reading Chemical Structures and Molecular Editor sections of this manual. If for example the query molecule you want to search is in a chemical spreadsheet you can right click on chemical in the spreadsheet and select Query Molecule.
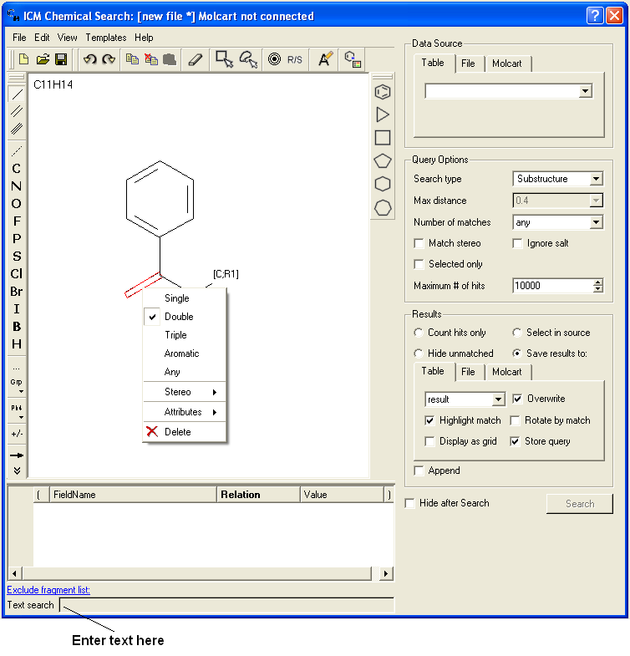
At this point your query can be modified as described in the Molecular Editor sections of this manual. However, there are a number of ways to specifically modify your query and filter your search. The way to accomplish this is to right click on a bond or atom and a menu as displayed below will be displayed. The menus differ depending on whether you right click on a bond or atom.
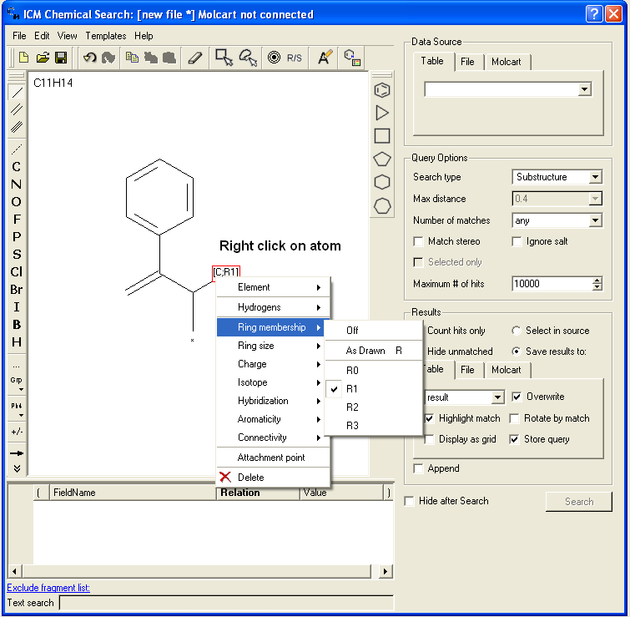
If you wish to specify a filter at an atom.
- Right click on the atom and the menu shown below will be displayed.
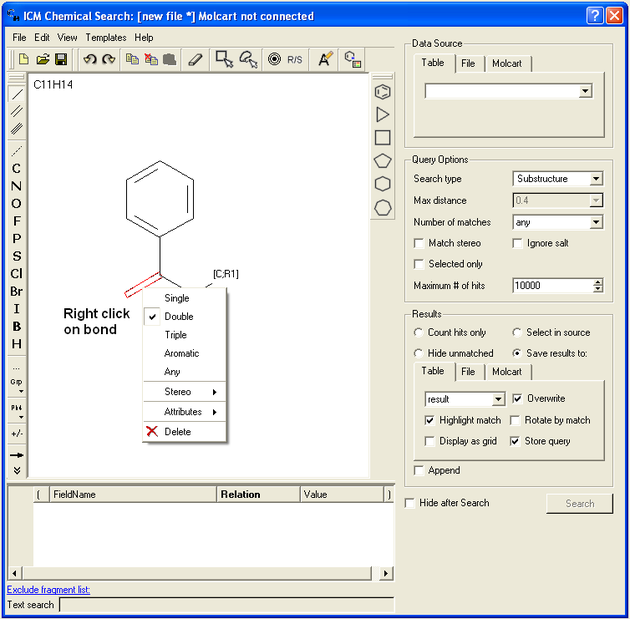
If you wish to specify a filter at a bond.
- Right click on the bond and the menu shown below will be displayed.
To specify a particular atom type, aromatic, aliphatic or R-group at a particular atom site.
- Right click on the atom and select the "symbol" option as shown below.
- Select the desired atom type, aromatic, aliphatic or R-group and a symbol will be displayed as shown below.
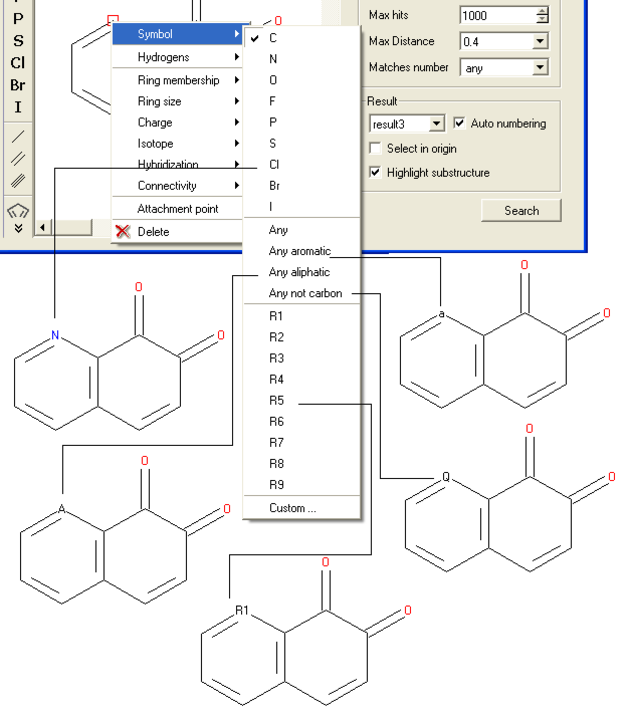
For example:
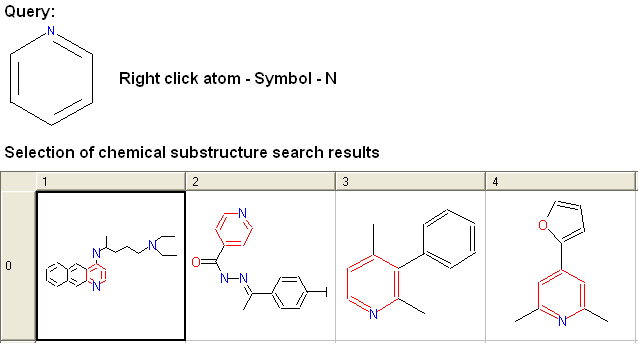
To specify a particular number of hydrogen atoms at a particular site:
- Right click on the atom and select the "Hydrogens" option as shown below.
- Select how many hydrogens you wish to specify and a symbol will be displayed as shown below.
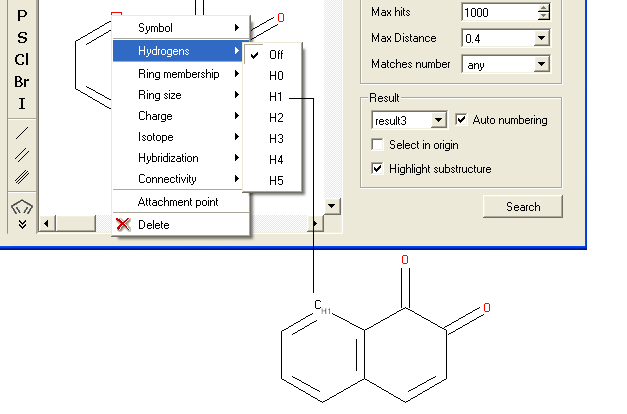
To specify the number of rings a particular atom will be a member of:
- Right click on the atom and select the "Ring membership" option as shown below.
- Select whether the atom should be part of 1, 2 or 3 rings.
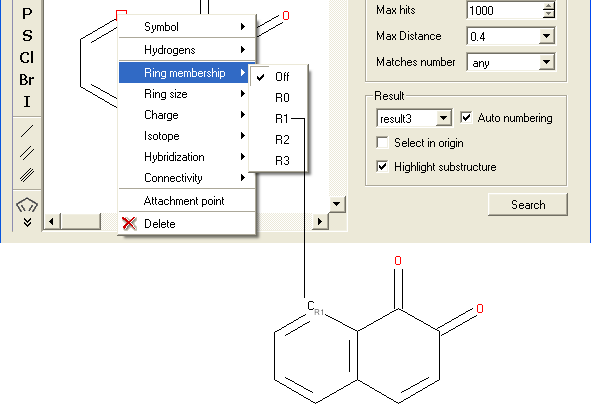
For example:
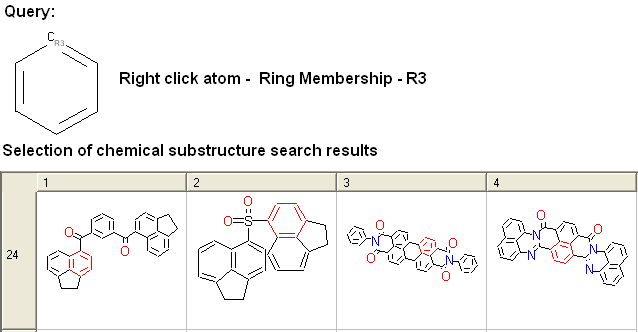
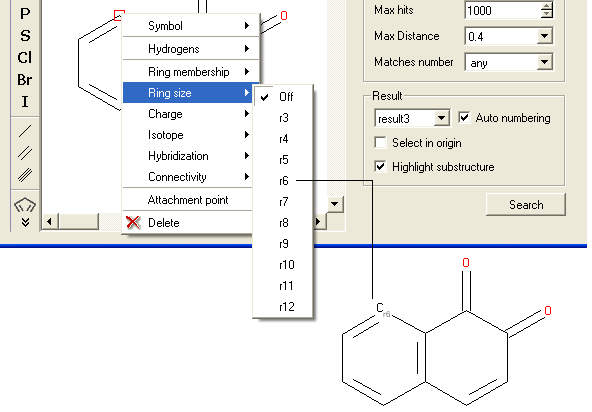
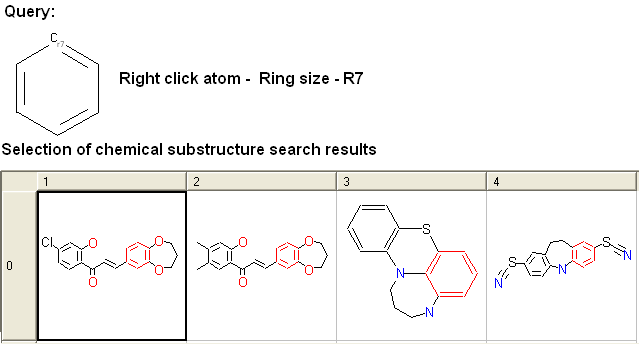
To specify the ring size connected to an atom:
- Right click on the atom and select the "Ring size" option as shown below.
- Select the size of the ring the atom should be connected to and a symbol will be displayed as shown below.
For example:
To specify the charge at a particular point:
- Right click on the atom and select the "Charge" option as shown below.
- Select the desired charge and a symbol will be displayed as shown below.
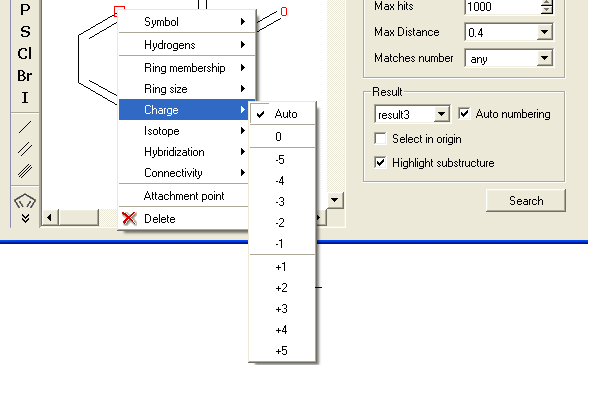
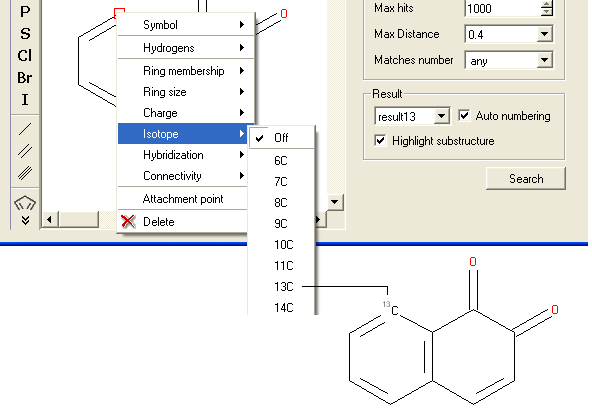
To specify an isotope at a particular atom
- Right click on the atom and select the "Isotope" option as shown below.
- Select the desired isotope from the list and a symbol as shown below will be displayed.
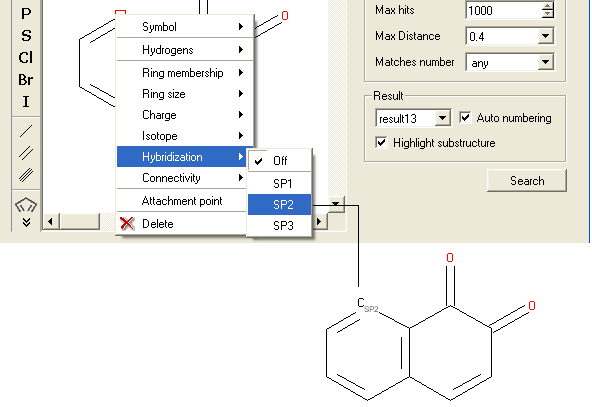
To specify the hybridization state of the atom:
- Right click on the atom and select the "Hybridization" option as shown below.
- Select the desired hybrization state and a symbol will be displayed as shown below.
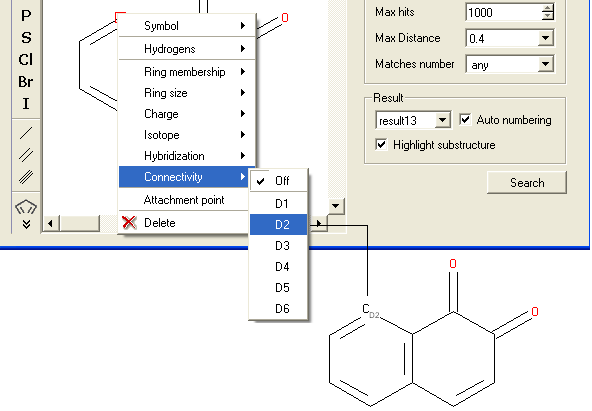
To specify the number of atoms you wish an atom to be connected to:
- Right click on the atom and select the "Connectivity" option as shown below.
- Select the number of atoms you wish the atom to be connected to and a symbol will be displayed as shown below.
To specify an attachment point - ie the position at which substituents will be added
- Right click on the atom and select the "Attachment point" option as shown below.
- An asterisk representing the attachment point will be displayed next to the atom.
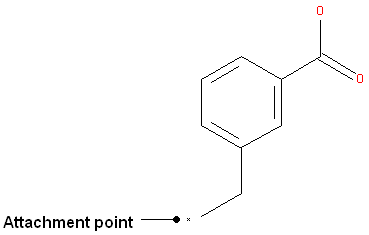
An attachment point means that the atom can be attached to zero or more bonds to heavy atoms.
10.5.2 Filter Search |
How to filter your query
To filter your query:
- Right click in the box shown below and select 'Add Condition'
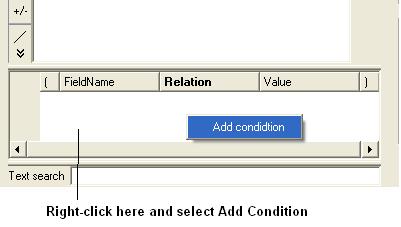
To add conditions to your filter:
- Double click in the fields labeled "Name" and "Relation" and select the options from the drop down arrow or type in values.
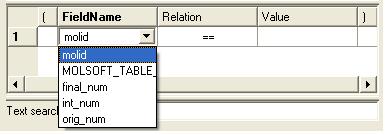
To remove a filter, right click on the filter and select 'Remove Filter'.
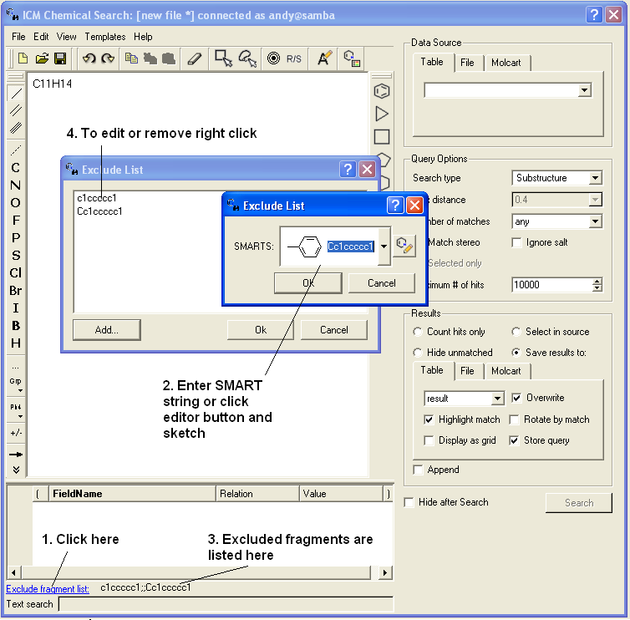
To exclude a fragment from your search
Click on the option Exclude fragment list:
- Enter the SMART string of the fragment or use the Molecular Editor option to sketch the fragment.
- Click OK and the fragment will be listed at the bottom of the chemical search window as shown below.
10.5.3 Query Processing |
To begin processing your query first you need to decide which database to search. The options are listed in the Data Source section of the ICM chemical search window.
Data Source
You can either search a Table- Chemical Spreadsheet a File - Local Database or MolCart.
If you select MolCart you first need to setup the link to the correct database.
- Enter the Server Name in which the database is stored.
- Enter the database name.
- Enter your username and password for the server.
- You can save these details so you do not have to re-enter this information each time you use the chemical similarity search.
If you are searching a Table, click on the Table tab and then select the drop down button where the names of your currently loaded tables are stored.
If you are searching a File click on the File tab and then locate your local table database file .molt or if it is already loaded into ICM you can locate it with the drop down button.
Query Options
Now select a search type:
- Click on the drop down arrow next to the "Search Type" option in the Query Options panel.
- Select the search type you want to use.
A substructure search is a search whereby only the defined molecule in the query will be searched against the database. Whereas, a FP similarity search which stands for fingerprint search enables any fingerprint within a structure to be searched for in the database. The similarity method calculates Tanimoto distance using chemical fingerprints (1. -most distant, 0. - closest). The similarity = 1. - distance.
The Max distance option is available for use with the FP search and the Matches number option is for use with the substructure search. The option you do not require based on your search method will be blanked out. A "Max distance" value of 0 means that the search will only identify matches exactly the same as the fingerprint - the default is 0.4. The "Matches number" option allows you to stipulate how many times within a structure in the database your query can be found.
Additional options include:
- Match stereo option. With this option chirality will be taken into account in substructure and similarity searches.
- Ignore salts.
- Keep ring membership.
- Search tautomers. If this option is selected ICM will use different tautomeric forms of the query chemical during the search.
If you make a selection of your query ICM can use that selection to search. How to make selections in the Molecular Editor are described here. Enter the Maximum number(#) of hits you would like returned.
Results
Before processing the query determine how you would like your results displayed in the Results section of the Chemical Search window.
Count hits only - this option will count the number of hits and display this number in a window once the searching has been completed.
Select in source - If you are searching a table you can select and highlight the query in the source table that you are searching.
Hide unmatched - Hide unmatched will hide the compounds that were not matched from view.
Save results to: - this option gives you the option to save the output results to a Table- Chemical Spreadsheet a File -Local Database or MolCart.
Append - this option will allow you to append to current table, file or Molcart database.
Search
- Click on the Search button to execute the search. You can choose to hide the window after the search.
10.5.4 Search a Database by Text |
To search a database by text enter the text you wish to search in the Text Search data entry box at the bottom of the ICM Chemical Search window.
10.5.5 Giga Search |
Read about the Giga Search method here.. If you have an ICM-Pro or ICM-Chemist license then you can run the search but the number of hits is limited. To get the full search you will need a GigaSearch license key.
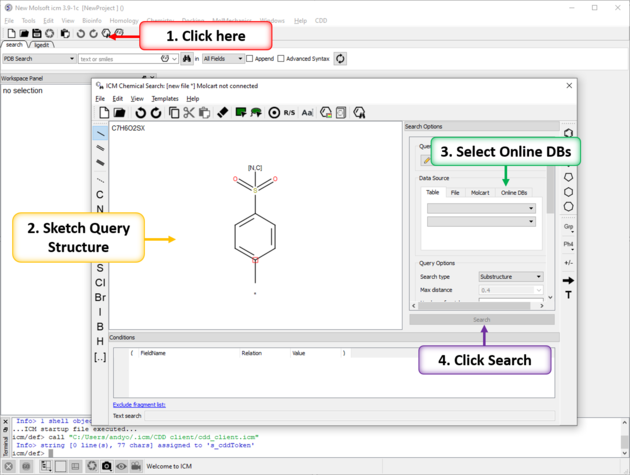
To perform substructure or similarity search of giga-sized libraries such as Enamine REAL or Chemspace FreedomSpace and others:
- Click on the chemical search button.
- Sketch your query chemical structure.
- Click on the Online DBs. tab in the data source window.
- Use the drop down button and choose Enamine REAL.
- Select query options.
- Click on the Search button.
10.5.6 Pharmacophore Drawing and Searching |
[ Pharmacophore Draw 2D | Pharmacophore Draw 3D | Pharmacophore Search | Extract 3D pharmacophore | Color 2D ]
Pharmacophores can be drawn in 2D in the ICM Molecular Editor or in 3D in the Graphical Display. 2D pharmacophore sketches can be used to search chemical tables (spreadsheets) containing 2D or 3D coordinates. A 3D pharmacophore can be used to search chemical tables containing 3D coordinates only.
10.5.6.1 Pharmacophore Draw 2D |
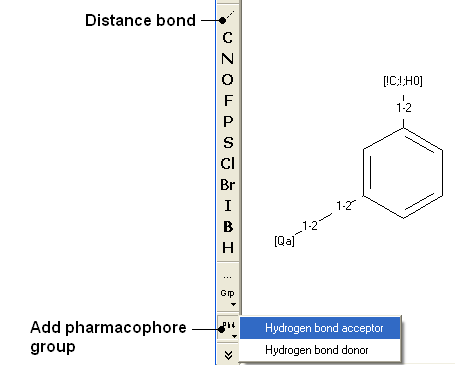
A 2D pharmacophore can be drawn using the ICM Molecular Editoror if you are going to use the drawing to search it is more efficient if you draw it in the Chemical Search window.
Use the distance bond button and the add pharmacophore group button to sketch the pharmacophore. The distance bond button represents the number of bonds between each pharmacophore point. You can edit the distance by right clicking on the bond and selecting edit. Other groups such as aromatic can be added using the standard molecular editor buttons.
| NOTE: Do not mix the 2D and 3D pharmacophore environment. For example do not edit a 3D pharmacophore in the 2D editor. |
10.5.6.2 Pharmacophore Draw 3D |
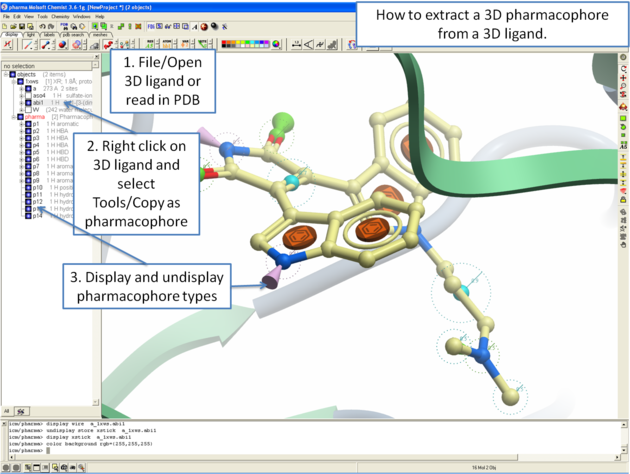
The easiest way to begin drawing a 3D pharmacophore is to draw a chemical in the ICM Molecular Editor which contains the key pharmacophore groups you want and then convert to 3D and extract the pharmacophore groups.
To draw a 3D pharmacophore this:
- Once the ligand is converted to 3D, right click on the ligand in the ICM Workspace.
- Select the Tools/Copy as Pharmacophore and choose the pharm centers option.
- The pharmacophore groups or centers will then be displayed in the graphical display and can be displayed and undisplayed in the ICM workspace.
To move a pharmacophore group:
- Use the drag atom button (picture of a hand. See below)
- Click on the pharmacophore group and then drag.
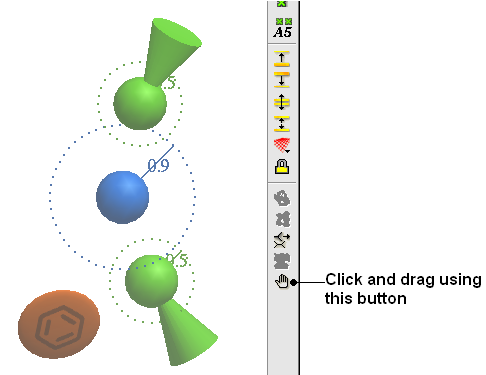
| NOTE: Distances between groups can be monitored using the atom distance measurement tool. See Calculating the distance between two atoms. |
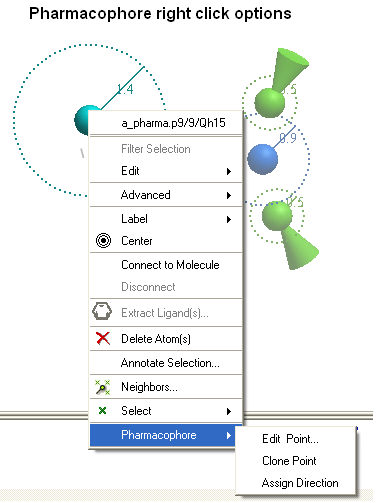
To change a pharmacophore group:
- Right click on the pharmacophore group in the ICM Workspace or in the Graphical Display.
- Select Pharmacophore/Edit Point
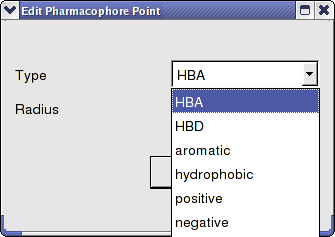
- Choose the group from the drop-down list shown below.
- Enter the desired radius.
To make a new pharmacophore group:
The easiest way to make a new pharmacophore group is to clone a pre-existing one. To do this:
- Right click on the pharmacophore group in the ICM Workspace or in the Graphical Display.
- Select Pharmacophore/Clone Point
- You can then move the new group as described above.
To change the direction of a pharmacophore group:
- Right click on the pharmacophore group in the ICM Workspace or in the Graphical Display.
- Select Pharmacophore/Assign Direction
- You can then move the new group using the drag atom button described above.
To remove the direction of a pharmacophore group:
- Right click on the pharmacophore group in the ICM Workspace or in the Graphical Display.
- Select Pharmacophore/Remove Direction
10.5.6.3 Pharmacophore Search |
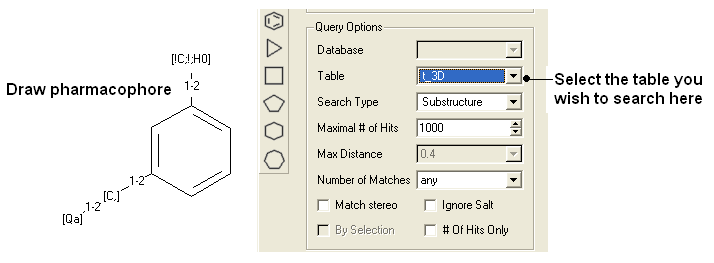
To perform a pharmacophore search using a 2D pharmacophore:
- Draw the 2D pharmacophore as described earlier in the Chemical Search window.
- Read in a molecular table to search or search a table in MOLCART.
- Select the chemical search options as shown below.
- Once the search has completed a new table with the results will be displayed.
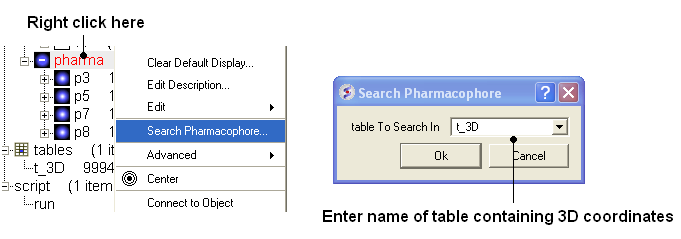
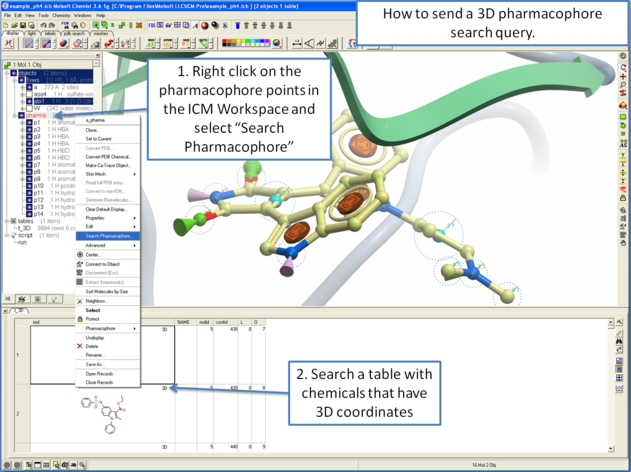
To perform a pharmacophore search using a 3D pharmacophore:
- Right click on the pharmacophore in the ICM Workspace
- Select Search Pharmacophore
- Use the drop-down button to select the table you wish to search. The table must contain 3D coordinates.
10.5.6.4 How to extract a 3D pharmacophore from a ligand. |
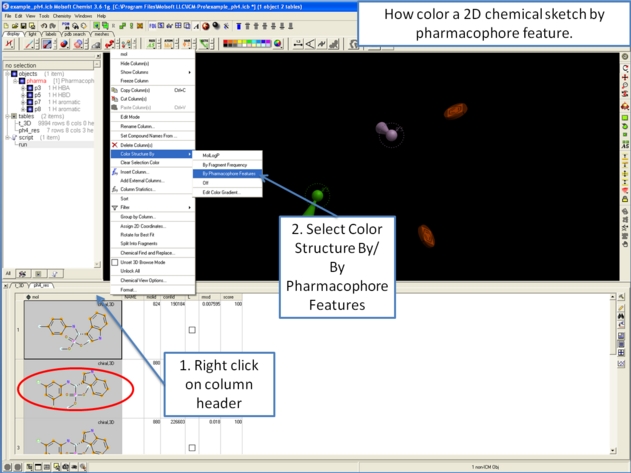
10.5.6.5 How to color a 2D chemical sketch by pharmacophore feature. |
| Prev Isis | Home Up | Next 2D Interaction Diagram |