[ Chemical clustering | Center | Re-order and Distance | Edit tree | Auto Close | Max Common Substructure | R-Group Decomposition ]
| Available in the following product(s): ICM-Chemist | ICM-Chemist-Pro | ICM-VLS |
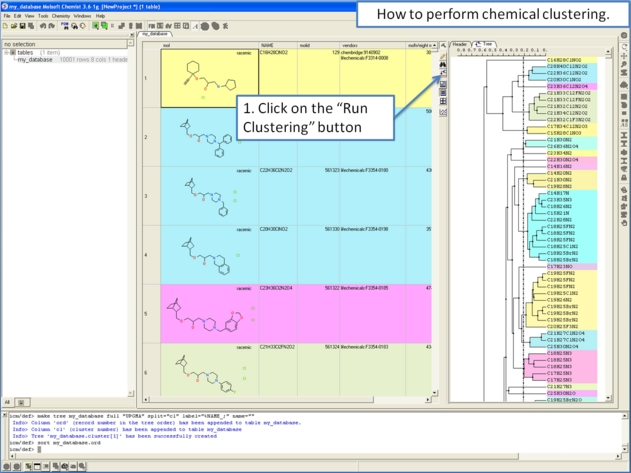
Clustering is described in more detail in the Tables Clustering section of this manual. To undertake chemical clustering choose:
- Chemistry/Cluster Set
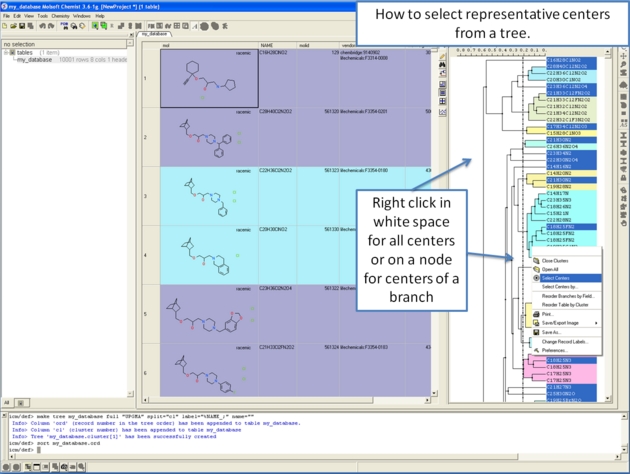
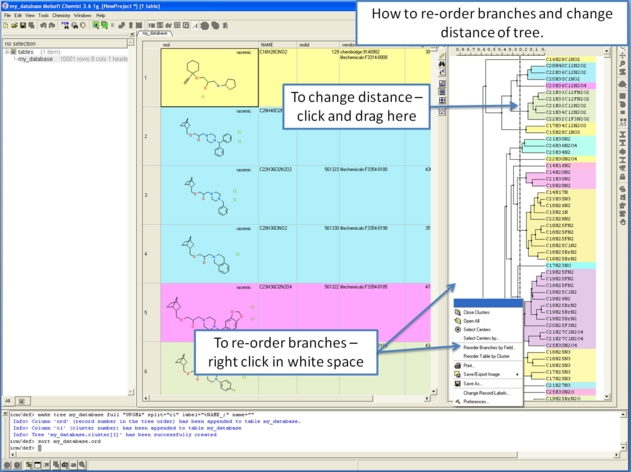

To make working with large cluster trees a little easier you can activate the auto close mode which will close downstream clusters and make them more compact.
To do this:
- Right click in white space on the cluster and choose Preferences.
- Select "Auto Close Clusters Downstream".
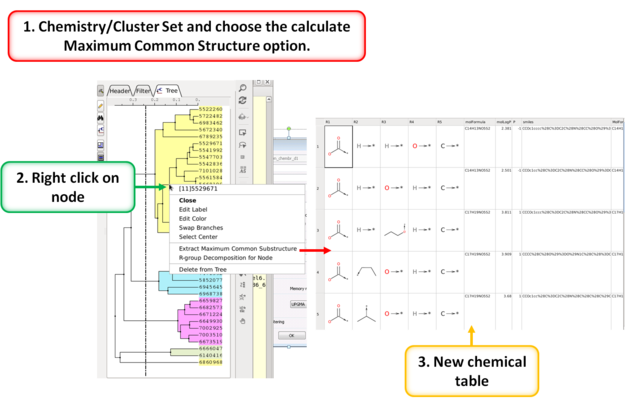
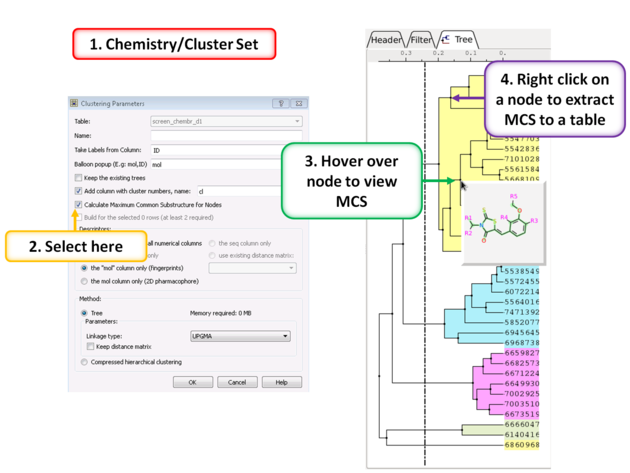
To calculate the Maximum Common Substructure (MCS) for nodes in a tree:
- Chemistry/Cluster Set
- Select the option Calculate Maximum Common Substructure for Nodes
- Hover over a node to view the MCS.
- Right click on a node and choose "Extract Maximum Common Substructure" to extract it to a table.
To perform R-group decomposition directly from a cluster node:
- Chemistry/Cluster Set
- Select the option Calculate Maximum Common Substructure for Nodes.
- Hover over a node to view the MCS.
- Right click on a node and choose "R-group decomposition for node".
The resulting table can be used for R-Group Enumeration or SAR.