| Available in the following product(s): ICM-Pro | ICM-VLS | ICM-Chemist-Pro |
The Atomic Property Fields (APF) superposition/alignment method was first reported by Maxim Totrov PhD (Principal Scientist - MolSoft) at the 2007 233rd American Chemical Society National Meeting, Chicago, IL USA and then published here.
APF is a 3D pharmacophoric potential implemented on a grid. APF can be generated from one or multiple ligands and seven properties are assigned from empiric physico-chemical components (hydrogen bond donors, acceptors, Sp2 hybridization, lipophilicity, size, electropositive/negative and charge).Here we describe template APF superposition whereby the APF is generated from a single or multiple template and is then globally optimized with the internal force-field energy of the ligand.
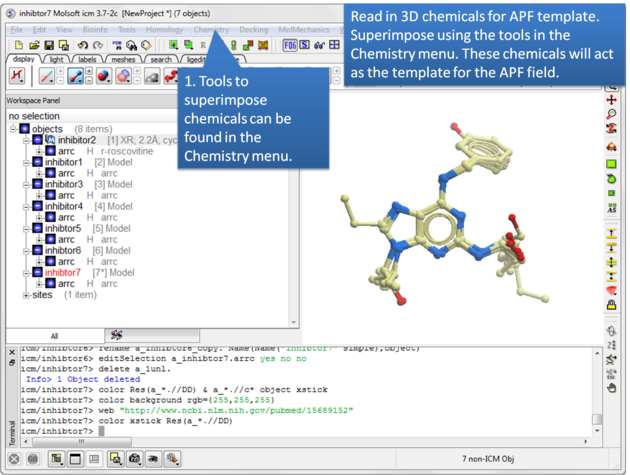 |
| Step 1: Read in the chemicals that will make up the Atomic Property Field you wish to dock to. |
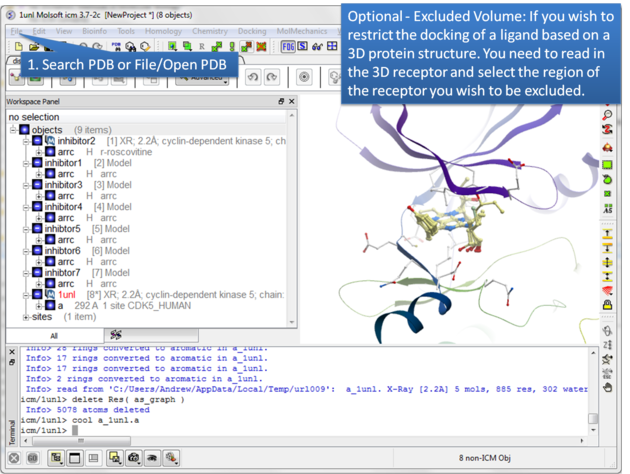 |
| Step 2: Certain regions of the pocket can be excluded from docking by selecting an excluded volume. |
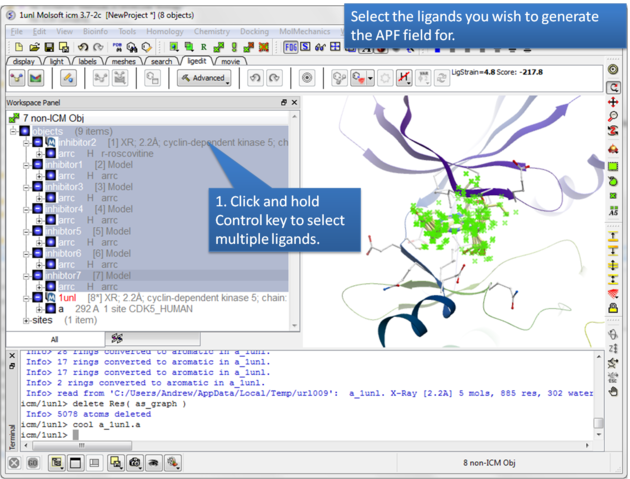 |
| Step 3: Select the ligands you wish to generate an APF field for. |
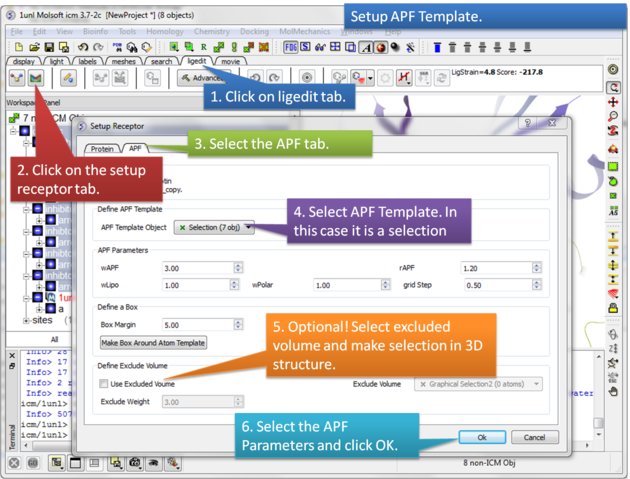 |
Step 4: Setup the APF template. The APF parameters are:
|
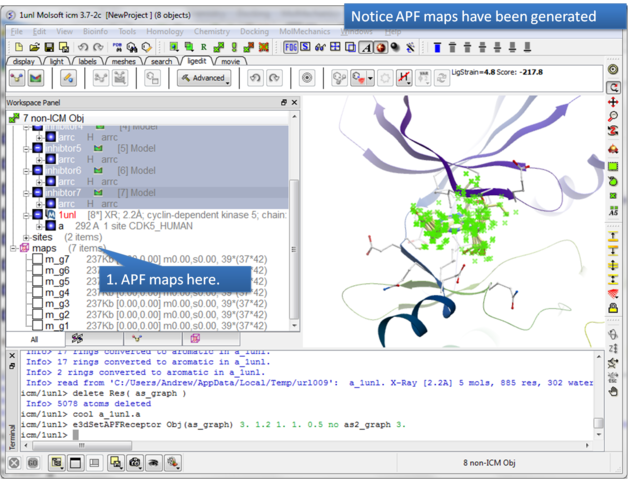 |
| Step 5: The APF maps are listed in the ICM Workspace left hand side. |
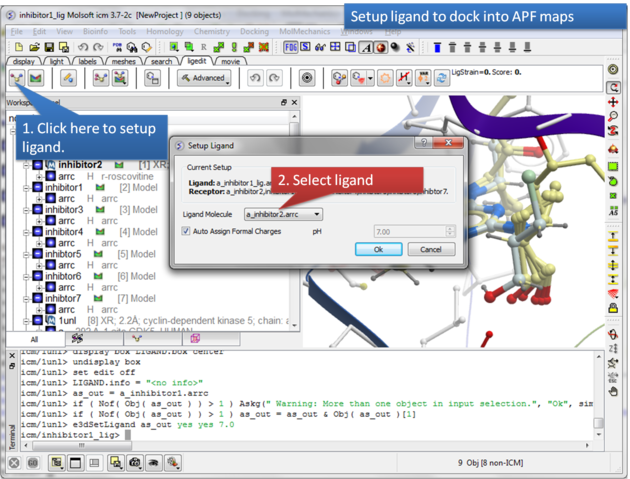 |
| Step 6: Select the ligand you wish to dock into the APF maps. |
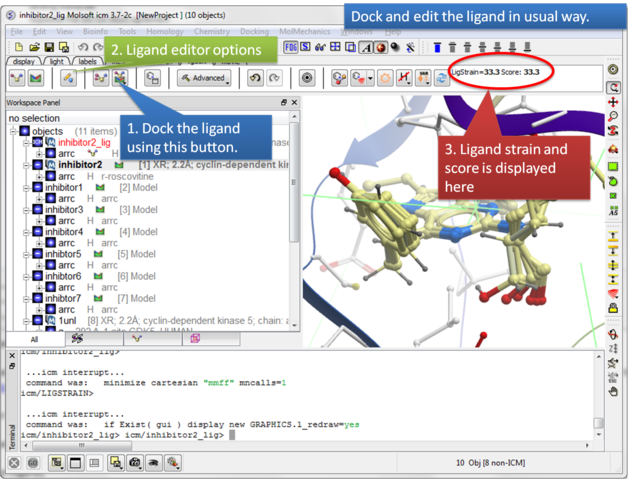 |
| Step 7: Dock the ligand using the ligand docking button and note the Score. |
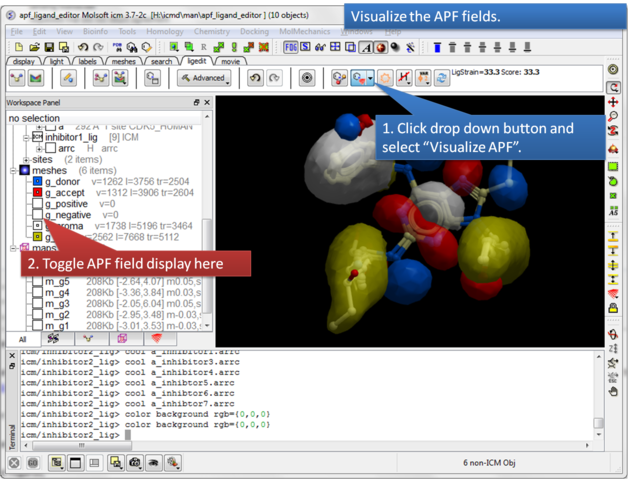 |
| Step 8: Visualize the APF maps. |