[ Convert PDB Chemical | Covalent bond | Write PDB | Renumber | Merge | Superimpose | RMSD | RMSD tips | delete residues | Atom distance | Ligand Pocket | Pocket conservation | Mutate Residue | Mutate Residue | Histidine Tautomer | Faq change torsion | Make Disulfide | read NMR | Solvent Accessible Area | weak hydrogen bonds | Formal Charge | Closest Neighbor ]
Questions and answers relating to protein and DNA structures, objects and the PDB
- How do I make a covalent bond between a ligand and a receptor?
- How do I convert a Chemical from the PDB into an ICM object?
- How to write a pdb file?
- How do I renumber the residues in a PDB file
- How can I merge two separate objects into one?
- How do I superimpose two proteins?
- How can I calculate the RMSD between two protein structures?
- Can you give me some tips on which options to use for RMSD calculations?
- I would like to delete all the residues in my protein except for the ones surrounding the ligand binding pocket.
- How do I display the distance between two atoms?
- How do I display only the residues that surround the ligand binding pocket?
- How do I show the sequence conservation around the ligand binding site?
- How do I mutate a residue?
- How do I mutate a terminal N or C residue?
- How do I change the tautomeric form of Histidine in a structure?
- How can I change the torsion angle?
- How do I make a disulfide bond?
- How do I read in all the structures in a PDB file of a protein solved by NMR?
- How do I write a script to calculate solvent-accessible surface and tabulate the results to show area for each residue in a protein?
- How do I display weak hydrogen bonds?
- How do I set a formal charge?
- How can I select the closest residue from the center of mass of a selected residue?
Please see the section in the cheminformatics chapter entitled Converting a Chemical from the PDB.
To make a covalent bond between a ligand and receptor:
Step one - place the ligand and receptor into the same object - how to move into one object is described here.
Step two - you can only make bonds in non-icm objects so you will need to "strip" the ICM object back to PDB Right click on the object name in the ICM Workspace and select Convert to non-ICM ...
Step three - use the ICM selection language to select the two atoms you would like to select and use the make bond command.
eg in 1f88 there is a covalent bond between the ligand retinal and K296 - zoom into it and see the bond - first we will delete this bond and then we will remake it like this:
delete bond a_1f88.aret/977/c15 a_1f88.a/^K296/nz
make bond as_graph
make bond a_1f88.aret/977/c15 a_1f88.a/^K296/nzIf you right click on an atom it will give you the icm selection language for each atom.
- Right-click on the molecular object name in the ICM Workspace Panel.
- Select -- Save as --
- Choose the file type
pdb
To renumber the residues in a PDB file you need to use the command line option
align number rs_residuesToBeRenumbered [ i_firstNumber ]
More details here:
http://www.molsoft.com/man/icm-commands.html#align-res-numbers
To merge two objects into one you can use drag and drop.
- Double click on the object you want to move in the ICM workspace (should be highlighted in blue).
- Click on the object you want to move and drag it to the name of the object (in the ICM workspace) you wish to move it to and then drop.

The quick way to do this is described in the section entitled "How to Superimpose Two Structures". However more superimpose options are found in the Tools/Superimpose menu.
- Display and select the proteins you wish to superimpose. You can select the whole protein simply by doublo-clicking on the name of the protein in the ICM Workspace. When selected there will be green-crosses on the protein in the graphical display and the protein molecules will be highlighted in blue in the ICM Workspace.
- Tools/Superimpose
There are three options
- Superimpose two proteins by 3D only
- Superimpose two proteins
- Superimpose multiple proteins.
In option 2 and 3 above you can select exactly what you would like to superimpose e.g. backbone, Calpha. See the image below. Also you can select which protein you want to remain static.

To calculate RMSD.
- First select the two molecules you wish to calculate the RMSD difference of. For example you can double click on the name of the ligand in the ICM Workspace (shaded blue when selected) and then whilst holding the CTRL key select the other ligand in the same way.
- Tools/Analysis/RMSD
- Select which parameters such as "Kept in Place" or "superimipose" and which atoms you wish to superimpose.
- The RMSD value will be displayed in the terminal window.
There are a number of ways to calculate RMSD and the method you use depends on the problem you wish to solve. For example you need to use different approaches when calculating the RMSD of proteins and ligands.
There are two options in GUI:
- Tools/Analysis/RMSD # For protein structures only. http://www.molsoft.com/gui/rmsd.html
- Superimpose Button # will work for chemicals and protein structures but beware the ligand is superimposed and therefore not useful for comparing docked structures http://www.molsoft.com/gui/superimpose.html
Command Line:
- RMSD command http://www.molsoft.com/man/icm-functions.html#Rmsd
- Static RMSD command # the way to compare two docked structures - use the chemical parameter and beware of how many atoms have been compared when comparing non-identical chemical structures. http://www.molsoft.com/man/icm-functions.html#Srmsd
You cannot delete from an ICM Object. You can delete from a model or PDB structure. So if the structure is an ICM object strip it to a model by right-clicking on the name of the object in the ICM Workspace and selecting "strip". Now follow these instructions.
- Double click (select) on ligand in the icm workspace and it will be colored dark blue and green crosses in the display
- Right click on the ligand in ICM workspace and select neighbors
- Enter 7 (Angstroms) and Type "Same Object Other Chains" - now you should see green crosses around ligand.
- If you want the whole residues surrounding the pocket select the Residue selection and you should see Rs rather than green crosses
- Now select the exclamation mark button which inverts selection - now everything but the surrounding residues are selected
- All selections are placed in the variable name as_graph.
- Type in the terminal window delete Res(as_graph)
One way to do this is to use the atom select button on the right hand side of the graphical user interface.
- The atom selection button has a green cross displayed on it see the selection buttons.
- Next select two atoms using this tool. Selected atoms will have green crosses on them in the graphical display.
- Select the labels tab (in older versions of ICM this is called the Advanced tab) and select the "Show distance between two atoms button".
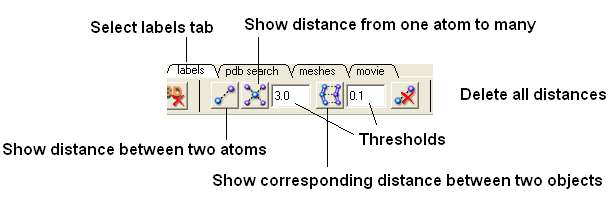
More instructions on how to display distances can be found in the section of the manual entitled "Finding the Distance Between Atoms"
There is a quick and easy way to do this as described in the Tips section of the manual entitled "Quick Binding Pocket Display" or you may want to follow the instructions below for a more user-defined method.
- Double click on the ligand name in the ICM workspace and it will be highlighted in blue.
- Right click on the name and select the Neighbors option.
- Enter 6 Angstroms (or whatever distance is appropriate for the ligand) for the sphere radius selection. Green crosses represent selected residues.
- Select type from the drop down menu "same_object_other_chains".
- Convert your selection to a residue selection by clicking on the propagate selection to all atoms in residue "R" selection button.
- Go to the display tab and select the representation you would like for the residues surrounding the pocket. Next use the "Invert Graphical Selection" button to select everything else other than the residues around the pocket and undisplay them by clicking on the representation buttons in the display tab.
See this tutorial.
- Display protein.
- Convert to ICM Object.
- Right click on the residue you wish to mutate. If you wish to mutate the C or N terminal please see the next question.
- Select Advanced/Mutate Amino Acid.
- Select amino acid from drop down menu.
For example we want to change the first and last residue of "1crn"
- Read pdb "1crn" or use the PDB search tab
- Convert pdb to an ICM object and select replace original - you can do this by right clicking on the name of the object in the icm workspace and select convert object.
- Extract sequence by right clicking on 1crn in the ICM Workspace and select extract sequence
- Right click on sequence and select edit sequence - double click on it and copy the sequence
- File New - select peptide and copy sequence into data entry box entitled one letter code
- Edit the first and last residue to the one you want
- Select ok and then you should have a long elongated unfolded peptide
- Type in terminal window
set tether a_2. a_1.- click on the advanced tab and select the button Toggle Tethers - to check tethers have been set - should see lots of red lines
- Type in terminal window
mncalls=10000minimize "tz"
- Display protein.
- Convert to ICM Object.
- Right click on the residue you wish to mutate. If you wish to mutate the C or N terminal please see the next question.
- Select Advanced/Mutate Amino Acid
- Select one of the following: Hip(histidine protonated), Hie (histidine epsilon tautomer)
See this section of the manual.
Use MolMechanics/Edit Structure/Set Disulfide Bond. More description of this can be found in the section entitled Making a disulfide bond.
- In the terminal window type the following.
read pdb pdb_FileName alle.g.
read pdb 1sgg all
Here is a script to calculate solvent-accessible surface and tabulate the results to show area for each residue in a protein:
read pdb "1crn"
show surface area mute # compute surface areas
res = a_/1:18 # residue range of interest
n = Nof(res) # the number of residues.
add column t Sarray(n, Name(Obj(res))[1]),Trim(Label(res),all),Area(res)
write t_1 separator="," "t.csv" # read into Excel or something else similar
To display a weak hydrogen bond you may have to change the ICM parameter which controls the hbond strength threshold for hbond display. This parameter is called:
GRAPHICS.hbondMinStrengthBy default it is set to 1 but the strength value can be set between 0. and 2. e.g By changing 1. to 0.2 you will see more weak hydrogen bonds.
To set a formal charge:
- Display the molecule
- Simply right click on the atom and choose Edit---> Set formal charge.
See the ICM language manual here :
http://www.molsoft.com/man/icm-functions.html#select-by-center-of-mass