| Prev | 3.4 Sequence, searches and alignments | Next |
[ sp | spf | bindingsiteanalysis | spt | prl | pdbmerge | ssd | sph ]
3.4.1 How to search all Prosite patterns in your sequence | [Top] |
read pdb "2dhf" make sequence a_1.1 # sequence of a PDB structure show sequence find prosite 2dhf_a # 2dhf_a is the sequence of the proteinSee also find prosite, find pattern and read prosite.
3.4.2 How to find a fragment in the PDB database ( obsolete ) | [Top] |
To understand the meaning of the arguments, see the find pdb command.
Examples:
read object s_icmhome+"crn"
call s_icmhome+"_qsearch"
# no graphics, just the list of solutions
# qsearch a_/2:6,14:18
# interactive
iqsearch a_1crn./2:6,14:18 "xxxxx------xxxxxx" "*" "*" .7
3.4.3 How to identify binding pockets | [Top] |
There are three algorithms (A, B, and C) with ICM which can identify pockets:
| option | target | macro |
|---|---|---|
| A | closed pockets | icmCavityFinder |
| B | almost closed pockets | make map potential , etc., see below |
| C | pockets with good ligand-binding potential | icmPocketFinder |
Example:
read pdb "1a28" delete a_!1,2 convert delete a_2 icmPocketFinder a_ 3. |
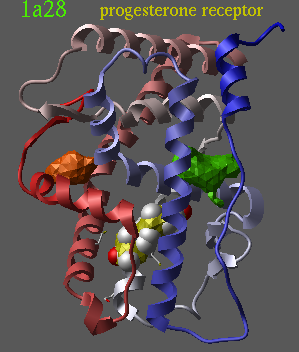
|
In the following example we find an almost closed pocket which can not be identified with icmCavityFinder .
read pdb "1fm6" # read the 'a' chain of RXR delete a_!1,9 # keep the RXR and its ligand only make map potential a_1 Box( a_ 1. ) 1. # grid size 1.5 A make grob m_atoms exact 0.1 solid split g_atoms cool a_ display g_atoms2 reverseIf you have problems with identifying pockets, change the grid size, the threshold level for make grop m_atoms , or try to convert object to the ICM type (the conversion will add hydrogens and make the object more dense).
3.4.4 How to find a similar fold or topological motif in the PDB database | [Top] |
read object s_icmhome+"crn" searchObjSegment a_1.1 30 3. # or read pdb "1pxt" delete a_!1 convert searchObjSegment a_1.1 24 6.You may need to adjust the seed fragment length and the RMSD parameters for a cleaner list.
The database foldbank.seg is provided and may be recompiled, customized and updated by the supplied _mkSegmentLib script.
See also segment, find segment, write segment, foldbank.seg, How to extract a diverse set of PDB entries How to compile a database of protein secondary structures and their folds .
3.4.5 How to generate a non-redundant list of PDB sequences | [Top] |
l_commands=no
errorAction="none" # if something goes wrong do not
# interrupt the loop
s_pdbDir = "/data/pdb/" # make sure you have correct path
pdbDirStyle = 4 #
read sarray s_pdbDir+"/derived_data/index/source.idx"
# you need a list of all pdb-entries
# (4 char. code per line will do)
source = Tolower(Trim(Field(source,1)))
n=Nof(source)
for i=1,n
read pdb sequence resolution source[i]
# append resolution to the chain name (like 9lyz_a19)
endfor
group sequence "*" uniqSeqs unique 0.1
# cutoff inter-sequence
# distance 0.1 (dissimilar by more than 10%)
#
# Other possibilities
#
# group sequence uniqSeqs unique 5 # if two seqs differ by more
# # than 5 mutations
# group sequence uniqSeqs unique # throw away only identical
# # sequences
#
delete sequences # get rid of sequences not
# included in uniqSeqs
write sequence s_inxDir + "/pdb1.seq"
# actual sequences for searches
write Name(uniqSeqs) "chainList"
# list of protein chains if you need it
quit
3.4.6 How to merge several pdb files | [Top] |
read pdb "1crn" read pdb "1d48" move a_2. a_1. # merges objects write pdb a_1. "both" # saves both files in pdb format write object a_1. # saves merged object in compact binary form
Before or after merging, the objects can also be edited, translated to a new position, rename chains, change residue numbers etc. Example:
read pdb "1d48" delete a_w* delete a_2 # delete the second chain read pdb "1crn" delete a_/33:99 # delete a C-term. part of crambin move a_1. a_2. # merge the remains write object a_
If you want to re-engineer a polypeptide chain of a protein, using two pdb-files, e.g. to transplant one part of a protein to another and restore the bonding connectivity, you may use the modify command:
read pdb "1crn" # one pdb read pdb "1cbn" # similar protein modify a_1./20:25 a_2./20:25 # translants a loop from 2nd object to the 1st one write pdb a_1. "combo"
3.4.7 How to compile a database of protein secondary structures and their folds | [Top] |
l_commands =no
l_info =no
l_confirm =no
errorAction="none"
segMinLength =3
mncalls =300
s_icmhome ="./"
s_reslib ="icmGAP" # Gly-Ala-Pro residue library
read library
# ...getting the representative list of chains...
read sequences s_pdbDir+"/derived_data/pdb_seqres.txt"
#make sure to have _mkUniqPdbSeqs executed recently
li=Name(sequence)
delete sequences
#...you may modify the method or create your own list...
if (Error) quit
unix mv foldbank.db foldbank.db.OLD
unix mv foldbank.seg foldbank.seg.OLD
for i=1,Nof(li)
lii=Tolower(li[i])
read pdb lii[1:4]+"."+lii[6]+"/"
delete !Mol(a_*/A) # delete HET-molecules
convert
er=r_out
rz=Resolution(a_1.)
if(rz < 0.01)rz=9.99
sx=Sstructure(a_*)
assign sstructure
# uncomment the following line, if you'd like
# to save GAP objects. requires GAP subdirectory
# write object "GAP/"+lii[1:4]+lii[6]
sprintf "# %d\nNA %s.%s\nRZ %.2f\nER %.3f\nSE %s\nSX %s\nSS %s\n" \
i lii[1:4] lii[6] rz er String(Sequence(a_*)) sx Sstructure(a_*)
write append s_out "foldbank.db"
assign sstructure segment
rename a_2. lii[1:4] # restore the original pdb-name
write append segment "foldbank.seg"
delete a_*.
endfor
quit
3.4.8 How to search headers of the PDB entries | [Top] |
- In unix: grep -i kinase PDB.tab
- From ICM: you have more possibilities:
read table s_icmhome+"data/inx/PDB.tab" # or in s_userDir+"inx/" show PDB.head ~ "kinase" # or show PDB.head ~ "*kinase*" # or show PDB.comp ~ "kinase*" # regular expressions # You can also a=PDB.head~"kinase" for i=1,Nof(a) nice PDB.ID pause delete a_*. endfor
- Use the gui (Find.In PDB.By Keyword..)
| Prev cavityanalysis | Home Up | Next ez25 |