| Prev | 3.3 Structure analysis | Next |
[ sht | a3ht | rht | hht | ir | htselres | it | pd | pca | dac | att | hp | cavityanalysis ]
3.3.1 How to optimally superimpose two 3D structures | [Top] |
|
Optimal superposition implies optimization of the Ca-RMSD upon
rigid body superposition of the equivalent residues/atoms.
This set of equivalent positions can be predefined, or determined
by sequence alignment, or automatically derived from structure.
In the latter case the optimized value is
the RMSD of a trial alignment is corrected by the alignment length
to reward longer alignment with slightly worse RMSD.
There are several different algorithms which can be applied: |
|
- superimpose by known residue or atom equivalences (see the superimpose command)
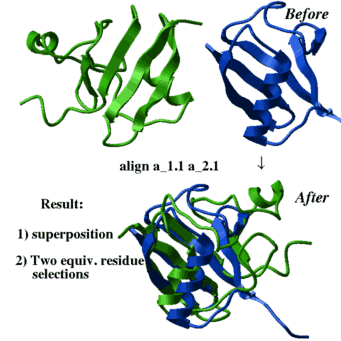
- superimpose by sequence alignment which is calculated on the fly:
superimpose a_1.1 a_2.1 align # the sequences are generated on the fly
This procedure fails if two structures have no significant sequence similarity - align .. command to find global structural alignment (returned by the ali_out variable)
and superimpose accordingly.
Here we do not rely on sequence alignement (although it can be added to
the optimized scoring function with a certain weight).
align a_1.1 a_2.1
- Align(.. distance) for local structural alignment
Use superimpose command. It performs an optimal rotation and translation of one structure onto the other. If necessary, a sequence alignment may be done prior to superposition by specifying align option in the command line.
Example:
read pdb "3znf"
display a_1.1//n,ca,c magenta
make sequence a_1.1
read pdb "1ard"
display a_2.1//n,ca,c blue
make sequence a_2.1
show sequence
# somewhat different sequences of two Zn-fingers
# sequence alignment is required
superimpose a_1. a_2. align
Note, in this particular example, the whole structural similarity
is not so high. However, better fit may be obtained if only portions
of the structures are superimposed, for example:
superimpose a_1.1/16:27/n,ca,c a_2.1/116:127/n,ca,c alignSee also: Rmsd( ) and Srmsd( ).
3.3.2 How to optimally superimpose without the residue alignment | [Top] |
align ms_molecule1 ms_molecule2
command. There are two variants: a fast superposition using dynamic programming algorithm align [distance] ms_1 ms_2 or a more rigorous, but somewhat less stable and slow align heavy ms_1 ms_2 ... command. This first command is well described above and identifies only the best superposition. The initial superposition is then refined similarly to the find alignment command.
The second algorithm (option heavy ) identifies a number of possible superpositions (solutions) based on the Ca atom coordinates only. The first solution is the best hit. See also load solution command.
Examples:
read pdb "4fxc" read pdb "1ubq" display a_*.//ca,c,n color molecule a_*. align a_1.1 a_2.1 center color red as_out color blue as2_out show ali_out
3.3.3 How to make a Ramachandran plot | [Top] |
plotRama rs_selection l_addLabel l_addBoundaries
Important: if a PDB structure is analyzed, convert it first to get a proper ICM-object (true ICM-molecular object does not require prior preparation for building Ramachandran plot).
Example:
read pdb "1crn"
convert a_1. # Note, one more object appeared in addition
# to the original (PDB) object 1crn
l_addLabel = yes # add residue labels to the plot
l_shadedBoundaries = yes # add allowed regions to the plot
plotRama a_2. l_addLabel l_shadedBoundaries
quit
3.3.4 How to display hydrogen bonds | [Top] |
The display hbond command allows to show the deviation angle of the hydroben bond from linearity (see the GRAPHICS.hbondStyle preference ).
Examples:
read object "crn" # already converted
show energy
display
show hbond 2.5 a_/1:15 # list of H-bonds with H-X distance < 2.5 A
# appears in the text window
display hbond 1.9 # H-bonds shorter than 1.9 A are shown
GRAPHICS.hbondStyle = 3
display hbond
See also: GRAPHICS.hbondStyle
3.3.5 How to identify atoms or residues at the molecular interface | [Top] |
read object "complex"
display a_1,2//!h* # display both molecules
# without hydrogens
color a_1 yellow
color a_2 green
show area surface a_1//!h* a_1//!h* # calculate surface of
# the 1st molecule only
color red Sphere(a_2//* a_1//* 4.) & Acc(a_1/*)# interface residue atoms
# of the 1st molecule
# in 4 A radius vicinity
# of the 2nd molecule
show area surface a_2//!h* a_2//!h* # calculate surface of
# the 2nd molecule only
color blue Sphere(a_1//* a_2/* 4.)&Acc(a_2//*) # interface residue atoms
# of the 2nd molecule
# in 4 A radius vicinity
# of the 1st molecule
If, instead, you need to mark residues,
convert the selection of the interface atoms to residues with
the Res () command:
.... # same as above. .... resAtInterf = Res( Sphere(a_2//* a_1//* 4.)) & Acc(a_1/*) display residue label resAtInterfNote that in the Sphere command it does not matter if you specify the atom selection or a residue selection as an argument, since the function operates at the atom level anyway. The difference in the specification of the ICM selection in these two examples (usage of two slashes for atom selection, and one slash for residue selection):
Sphere(a_1.1/* 4.) versus Sphere(a_1.2//* 4.) and also Acc(a_1.1/*) versus Acc(a_1.2//*) for specifying residues and atoms, respectively.
Important: when calculating surface, be sure that you properly specify the selection arguments in the show area surface command.
3.3.6 How to select accessible, buried, hydrophobic, residues. | [Top] |
read pdb "1crn" delete a_W # delete water molecules show surface area # compute exposed surface areas for each residue show Acc( a_/* , 0.25 ) # show all residues exposed more than 25%) show !Acc(a_/* , 0.25 ) # show buried residues
Converting the selection into other formats
You can also show the selection in a different format, e.g.
String( Acc( a_/* )) # or a_1crn.m/1,5:8,11:12,14:15,17:20,22:25,28:29,31,33:34,36:46 Label( Acc(a_/*)) #>S string_array T1 P5 S6 I7 V8 S11 ...
Identifying buried polar residues
About 50% of all residues have relative accessibility less than 25%. Polar residues typically do not like to be buried. The charged residues like it even less, and if they are buried they usually form a salt bridge. Example:
read pdb "1qau" # neuronal nitric oxide synthase display display xstick a_/62,121 # buried asp and arg form a bridge
To identify buried charged residues, use combine the previous selection of the buried residues with specific residue type selection, e.g.:
read pdb "1qau" delete a_W show surface area show a_/asp,glu,arg,lys & ! Acc( a_/* 0.15 )Here we used a more conservative threshold of 0.15 as the burial threshold. Feel free to modify the selection of residue types above to find other buried residues.
Identifying exposed hydrophobic patches
A similar technique can be used to identify hydrophobic patches:
read pdb "1qau" delete a_W show surface area show a_/leu,ile,val,met,trp & ! Acc( a_/* ) # buried ones show Acc( a_/leu,ile,val,met,trp ) # exposed hydrophobs
To find clusters of exposed hydrophobic residues, use the Sphere function. The Sphere function returns atoms, and they need to be converted to residues with the Res function.
exposed_hres = Acc( a_/leu,ile,val,met,trp )
for i=1,Nof(exposed_hres )
nbrs = Res(Sphere( exposed_hres[i] exposed_hres , 5.0 ))
if Nof( nbrs ) >= 2 show nbrs # show pairs of exposed hydrophobs
endfor
3.3.7 How to identify torsions at the molecular interface | [Top] |
read object "complex"
display a_1.1,2//!h* # display both molecules
# of the complex w/o hydrogens
color a_1.1 yellow
color a_1.2 green
show area surface a_1.1//!h* a_1.1//!h* # calculate surface of
# the 1st molecule only
a1=Sphere(a_1.2//* 7.) & Acc(a_1.1//*) # define the 1st molecule atoms
# belonging to the interface
# of the 1st molecule
# in 7 A radius vicinity
v1=V_1.1//S & a1 # identify standard geometry
# torsions of the 1st molecule
# belonging to the interface
color red Atom(v1) # color atoms which torsions
# belong to
# similar for the 2nd molecule
show area surface a_1.2//!h* a_1.2//!h*
a2=Sphere(a_1.1//* 7.) & Acc(a_1.2//*)
v2=V_1.2//S & a2
color blue Atom(v2)
3.3.8 How to calculate packing density | [Top] |
read object s_icmhome+"crn" asel = a_/5:15 show volume skin asel asel rskin = r_out vwExpand = 0. show volume surface asel asel rsurf = r_out print "skin volume = ", rskin, "; vw volume = ", rsurf print "packing density = ", rskin/rsurf
3.3.9 How to perform a principal component analysis | [Top] |
read sequences s_icmhome+"zincFing" # read sequences from file,
list sequences # see them, then ...
group sequence alZnFing # group them, then ...
align alZnFing # align them, then ...
a = Distance(alZnFing) # calculate a matrix of pairwise
# distances among them
n=Nof(a) # number of points
b=Disgeo(a) # calculate principal components
corMat = b[1:n,1:n-1] # coordinate matrix [n,n-1] of n points
eigenV = b[1:n,n] # vector with n sorted eigenvalues
xplot = corMat[1:n,1]
yplot = corMat[1:n,2]
# plot 2D distribution
plot xplot yplot CIRCLE display
3.3.10 How to calculate a dihedral angle | [Top] |
Example:
read pdb "1crn" # read it in convert # quickly convert into ICM-object show v_//phi,psi # just show all phi,psi's show v_/3:17/xi* # show chi angles of residues from 3 to 17 a = Value(v_//phi,psi) # create a real array of values of spec. torsionsYou can calculate a dihedral angle between any two planes defined by three atoms (for example two Phe rings) with the calcDihedralAngle macro. If this macro is loaded, you can do the following:
read object "crn" as_1 = a_/1/n,ca,c as_2 = a_/3/n,ca,c display a_//n,ca,c blue color as_1 magenta color as_2 green calcDihedralAngle as_1 as_2 print "dihedral angle 1(n,ca,c) and 3(n,ca,c) (deg) = ", r_outNote that the order of atoms in the selections as_1 and as_2 is determined only by the ICM-molecular tree (will be the same for a_//n,c or a_//c,n). Thus, any changes in the selections as_1 and as_2 not changing their content has no effect on the resulting dihedral angle:
as_1 = a_/1/ca,c,n as_2 = a_/3/ca,c,n calcDihedralAngle as_1 as_2 print "dihedral angle 1(n,ca,c) and 3(n,ca,c) (deg) = ", r_outSee also: Acos( ), Length( ), Sum( ), Vector( ).
3.3.11 How to print a table of the torsion angles | [Top] |
show V_//* # or show V_//phi*,psi*,omg* # or show V_//xi* # side chain torsionsA nicer formatted output (one line per residue) may be generated with macro printTorsions, for example:
read pdb "1crn" convert printTorsions a_/2:15Note, that you do not need to convert your molecular object if it is an ICM-object.
3.3.12 How to build a hydrophobicity profile | [Top] |
# define a hydrophobicity scale
hPhobInd = { 1.8, 0.0, 2.5, -3.5, -3.5, 2.8, -0.4, \
-3.2, 4.5, 0.0, -3.9, 3.8, 1.9, -3.5, \
0.0, -1.6, -3.5, -4.5, -0.8, -0.7, 0.0, \
4.2, -0.9, 0.0, -1.3, 0.0}
# make a macro
macro hPhobProfile seq_ i_windowSize
if (Type(i_windowSize)=="unknown") then
i_windowSize = Ask("Enter window size",windowSize)
endif
R_window = Rarray(i_windowSize,1./i_windowSize)
R_hphob = Smooth (Rarray(seq_,hPhobInd), R_window)
R_ruler = {0.,0.,10.,10.,0.,0.,0.,0.}
R_ruler[2] = Real(Length(seq_))
r_tic = 1./Sqrt(Real(i_windowSize))
r_tic = Integer(r_tic*100.0)/100.
R_ruler[5] = -7.*r_tic
R_ruler[6] = 7.*r_tic
R_ruler[7] = r_tic
R_ruler[8] = r_tic
print R_ruler
s_legend = {"Hydrophobicity plot","Sequence","Hydrophobicity"}
xplot = Count(1 Length(seq_))
yplot = R_hphob
psfilename = "hphob"
plot xplot yplot R_ruler s_legend grid="xy" display psfilename
delete R_window R_ruler r_tic
endmacro
# now, an example
read object "crn"
s = Sequence(a_)
hPhobProfile s 7
3.3.13 How to display and characterize protein cavities | [Top] |
Examples:
read object "crn" # or whatever
make grob skin "g_skin"
split g_skin
nShells = i_out
display wire residue labels
for i=1,nShells
v = Volume(g_skin$i) # actually its surface is returned in r_out
s = r_out # there is no need to use the explicit Area(g_skin$i)
if(v > 0.) then # note that cavities have negative volume!
display transparent smooth g_skin$i
printf "Shell %d: V=%f A=%f\n", i, v, s
else
display reverse smooth yellow g_skin$i
center g_skin$i
printf "CAVITY %d: V=%f A=%f R~%f\n", i, -v, s, -3.*v/s
endif
# add pause here for an interactive session
endfor
| Prev cbc | Home Up | Next sh24 |