| Prev | ICM User's Guide 17.2 Flexible Receptor Docking and Multiple Receptor Conformations | Next |
[ Fully-Flexible Docking | Multiple Receptor Conformation Docking ]
The standard ICM docking procedure desribed in the previous section incorporates a flexible ligand and a semi-rigid receptor wherby flexibility is incorporated by including soft van der Waals potential maps. However due to ligand induced-fit there is sometimes a need to incorporate flexibility more explicitly by allowing the side-chains of the receptor to be fully flexible or by using multiple-rec{multiple conformations} of a receptor in the docking procedure.
17.2.1 Fully Flexible Ligand and Receptor Docking |
To undertake fully-flexible ligand and receptor docking you first need to dock the ligand into the receptor using the procedures described in the previous section entitled Small Molecule Docking. The next step is to select the pose from your docking experiment you wish to use for flexible docking.
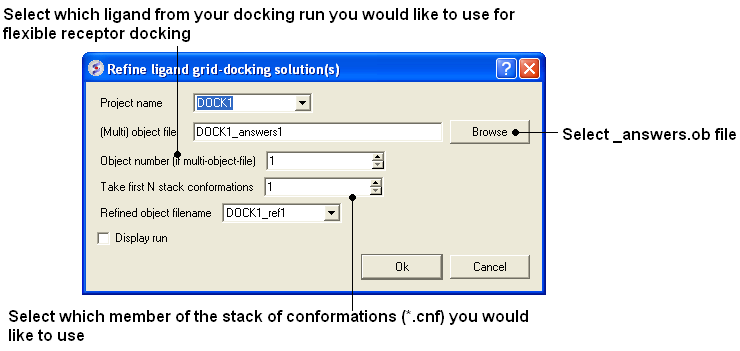
- Docking/Flexible-Receptor/Refinement and a window as shown below will be displayed.
- Enter the name of the initial docking project.
- Select the answers.ob file from the docking experiment.
- If you docked more than one ligands you need to select which ligand you want to use for the flexible receptor docking. For example if you want to refine the 3rd ligand in your docked database you would enter 3 in the Object number data entry box.
- Select which member of the stack of docking solutions you wish to use. In most cases it will be the first member of the stack and therefore you will enter "1".
- Enter a name for the refined complex.
- You can display the run in the graphical display but this will slow the process down.
17.2.2 Multiple Receptor Conformation Docking |
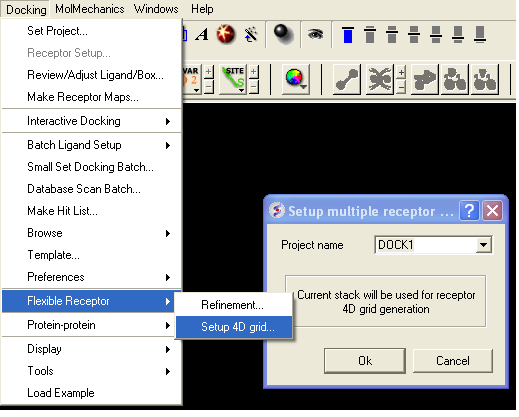
To dock to more than one conformation of a receptor you must first generate a stack of conformations. One way to do this is to select a number of side-chains and right click on the graphical selection and choose Advanced/Optimize Side Chains or right click on the ligand in the binding pocket and select Advanced/Optimize Ligand Vicinity. Other methods of generating a stack of conformations involve using the command line (see: http://www.molsoft.com/man/icm-commands.html#store-conf). A stack can be viewed using the MolMechanics/View Stack option and a table is generated which can be clicked on to view the different conformations. If there are too many elements in the stack redundant conformations can be deleted using the
The docking procedure is the same as described in the Small Molecule Docking section. The only difference is before generating the maps you need to select Flexible Receptor/ Setup 4D grid.
| Prev Reload | Home Up | Next Template Docking |