| Prev | ICM User's Guide 17.1 Small Molecule Docking | Next |
[ Receptor Considerations | Ligand Considerations | Setting up the Docking Project | Set Project Name | Setup Receptor | Review and adjust binding site | (Re)Make Receptor Maps | Begin the Docking Simulation | Interactive Docking | Batch Docking | Viewing Your Docking Results | Scan Hits | View Stack | Make a HitList | Reload ]
This section is concerned with predictions of interactions of drugs or small biological substrates (less than about 600-700 Da) to pockets of larger, more rigid, receptors (typically, protein molecules, DNA or RNA).For accurate ligand docking, the goal is to have an adequate three-dimensional model of the receptor pocket you are planning to dock ligands to. If this is the case then ICM docking has been shown to be very accurate in a number of independent assesments.
However, there are a number of pitfalls which need to be overcome to achieve accurate ligand docking. The pitfalls are that your model is not accurate overall, does not reflect the induced fit, or alternative conformations of the receptor binding pocket are missed.
Some facts about ICM docking:
- An average docking time is 20 seconds to 3 minutes per ligand per processor.
- ICM docking is probably the most accurate predictive tool of the binding geometry today. ICM ranked first place compared to other leading docking software in terms of accuracy in a recent analysis undertaken by Astra Zeneca scientists. See: On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. Hongming Chen, Paul D. Lyne, Fabrizio Giordanetto, Timothy Lovell, and Jin Li J. Chem. Inf. Model.; 2006; 46(1) pp 401 - 415
- The time per ligand was chosen to be the smallest possible to allow screening of very large data sets. To increase the time spent per ligand, change the Docking_thoroughness parameter.
17.1.1 Receptor Considerations |
If you have only a single PDB entry for your receptor, convert the protein to an ICM object, delete water molecules and irrelevant chains. However, if you have a choice between several templates, take the following into account:
- X-ray structure is preferable to an NMR structure
- High resolution X-ray structure ( less than 2.1A ) is much better than, say 2.5A . Watch out for high-B-factor regions and avoid them; sometimes crystallographers deposit fantasy coordinates with high-B-factors.
- Place polar hydrogens and choose correct form of histidine.
- A bound conformation of the receptor is preferable, however if you use an apo-model, an NMR structure or a model by homology, the side-chains in a pocket may be incorrect. Frequently they stick out and prevent a ligand from binding. Those stubborn side-chains can be 'tamed', (i) manually; (ii) by a side chain simulation with elevated surfaceTension; or (iii) by an explicit flexible docking calculation with a known ligand.
- A model by homology can be built with the build model command (see molecular modeling section of this manual) and used for docking.
17.1.2 Ligand Considerations |
Usually a good start is to try to dock the known ligand(s) to the receptor model. You may also want to dock a library of compounds in order to identify lead candidates. In this case the main pitfall is that the library is too restricted, molecules are not chemically feasible or not drug-like.
| NOTE: If you are docking a ligand directly from the PDB please check the bond types and formal charges of the ligand. This is discussed in the section entitled Converting a Chemical from the PDB |
17.1.3 Setting up the Docking Project |
ICM ligand docking procedure performs docking of the fully flexible small-molecule ligand to a known receptor 3D structure. The goal of the flexible docking calculation is prediction of correct binding geometry for each binder. ICM stochastic global optimization algorithm attempts to find the global minimum of the energy function that includes five grid potentials describing interaction of the flexible ligand with the receptor and internal conformational energy of the ligand. During this process a stack of alternative low energy conformations is saved (one of the choices in the Docking menu ). Before setting up the docking project, an ICM object of the receptor has to be created. In most cases, x-ray structure of the receptor is initially in the PDB format. Thus, it has to be converted to the ICM format. This process involves addition of the hydrogen atoms, assignment of atom types and charges from the residue templates (icm.res) and imposition of internal coordinates tree (icm-tree) on the original pdb coordinates. To convert a pdb structure into icm object is through GUI as follows:
- Load receptor pdb file into ICM by clicking File/Open/PDB.
- Convert loaded structure into an ICM object by clicking MolMechanics/ICM-convert/Protein.
|
NOTE: It is recommended that "optimize hydrogens" option is selected. To accelerate the procedure, disable the 3D graphics window (type in the terminal window |
Follow these instructions in order:
- Set Project Name
- Setup Receptor
- Setup Ligand Note:Version 3.4-7f and higher does not have this option - ligand setup is selected at interactive docking or batch setup step (See Start Docking Simulation)
- Review and Adjust Binding Site
- Make Receptor Maps
- Start Docking Simulation
- View Docking Results
17.1.4 Set Project Name |
Start the docking project setup by defining the project name:
- Click on Docking/Set project name
- Enter a unique name into the Project name data entry box. Avoid spaces and leading digits in the name. All files related to the docking project will be stored under names, which start from the project name. Most customized parameters will be saved in the table file under the project name as well:
- Click on the 'OK' button.
Now set up the receptor. Go to Receptor Setup
17.1.5 Setup Receptor |
The next step is to set up the receptor for docking.
- Click on Docking/Receptor setup
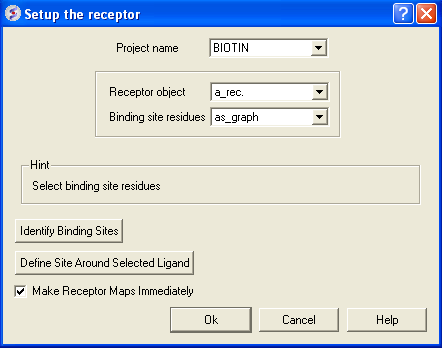
- Enter the project name in the Project name data entry box. If the project name was established in the same ICM session then it should automatically appear in this box.
| NOTE: Other docking project names that you have entered can be found by clicking on the arrow besides the Project name data entry box. |
- Enter the receptor molecule in the Receptor molecule(s) data entry box. In most cases a_* will do - all molecules in the current object will be included. The receptor molecule can also be found by clicking on the arrow next to the data entry box. A list of potential receptors will be displayed. Click on the receptor you wish to use for your docking experiment.
- Enter the ICM binding site residues Binding site residues data entry box. Define the binding site residues, either manually e.g. a_/123,144,152 for selection by residue numbers, graphically using the graphical selection tools such as the lasso tool (don't forget to set selection level to residue) or the icmPocketFinder function. If the residues are selected using the lasso tool or icmPocketFinder there should be green crosses surrounding the ligand binding pocket. The green crosses represent a graphical selection and are returned to a variable called
as_graph typeas_graph in the Binding site residues data entry box. This selection is used solely to define boundaries of the docking search and the size of the grids and doesn't have to be complete, selecting some 4 residues delimiting the binding site is sufficient. Receptor setup dialog also lets you run binding site identification routine to quickly locate putative binding sites on your receptor. - If you do not select the option Make Receptor Maps Immediately you can make the maps by using the option Docking/(Re)Make Receptor Maps.
| NOTE: Potential ligand binding pockets can be identified using ICMPocketFinder or by clicking on the Identify Binding Sites button in the Docking/Receptor Setup.. data entry window. These two methods for identifying pockets are identical. |
- Click on the OK button.
| NOTE: At this stage of the docking setup it is a good idea to keep an eye on the terminal window. Instructions and any error messages will be displayed in the terminal window. If you do not see the terminal window select Windows/Terminal Window. |
To complete the receptor setup there are two more steps:
Adjust the position of the probe (initial ligand starting position
The position of the probe (usually represented as 4 spheres in the center of the pocket) represents the initial position where sampling will begin. The default probe position is generally OK for most purposes but if you would like to move it to a critical part of the receptor so that sampling initially concentrates in that region you can do so using the middle mouse button and holding the SHIFT button for global rotation. Once you are happy with the position of the box press the enter key or click on "GO".
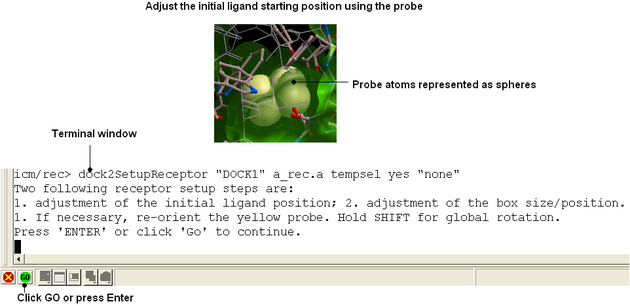
| NOTE The probe position can be changed again using the Docking/Review/Adjust Ligand/Box.. option. |
Adjust the size and/or position of the box The purple box represents the region in which maps will be generated. The box needs to be large enough to encompass the binding pocket but not too large and including regions of the receptor which are not relevant for the ligand to bind. If the binding site is correctly defined in the earlier Receptor setup then the default box size is usually fine. If it is necesary to change the box size you can use the left mouse button with the cursor at any corner of the purple box to change it.
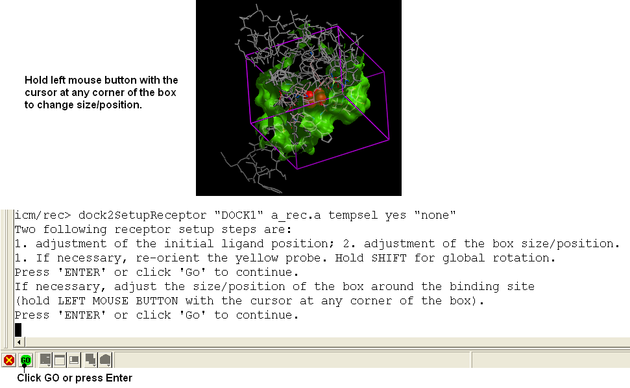
| NOTE The size of the box can be changed again using the Docking/Review/Adjust Ligand/Box.. option. |
17.1.6 Review and adjust binding site |
| NOTE: Generally the default box ICM generates in the receptor setup stage is adequate. It is usually a good idea to double check the box encompasses all the residues you want to dock to. |
ICM makes a box around the ligand binding site based on the information entered in the receptor setup section. The position of the box encompasses the residues expected to be involved in ligand binding, however you may wish to alter the size of the purple box or the position of the ligand probe (red spot).
- Click on the menu Docking/Review/Adjust Ligand/Box
- A data entry window will be displayed as shown below.
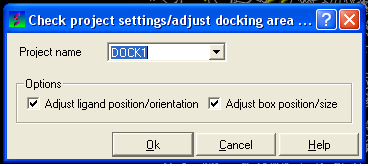
- Select the option Adjust/ligand position/orientation and/or Adjust box position/size
Follow the instructions in the command line display.
| NOTE: Always check that the correct project name is displayed in the data entry window. |
Now go to Make Receptor Maps.
17.1.7 (Re)Make Receptor Maps |
| NOTE: You need to use this option if you have changed the size of the box (Review/Adjust Ligand/Box). You also need to use this option if you did not select the Make Receptor Maps Immediately option in Docking/Receptor Setup. |
The next step is to construct energy maps of the environment within the docking box.
- Click on the menu Docking/Make Receptor Maps
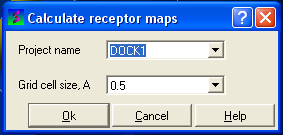
| NOTE: Always check the correct project name is displayed in the data entry window. |
- Select the resolution of the map by entering a value into the grid cell size data entry box. We recommend a value of 0.5 for both accuracy and speed of calculation.
| NOTE: Calculation of the maps may take a few minutes. |
Now begin the docking procedure.
17.1.8 Begin the Docking Simulation |
Once the receptor and maps have been correctly set up then the docking procedure can begin.
There are two options INTERACTIVE or BATCH docking (Please note some of the options may be limited for users without ICM-VLS)
17.1.9 Interactive Docking |
Use interactive docking to dock one ligand at a time in the foreground. It is ideal to use this option for small-scale docking.
- Click on the menu Docking/Interactive Docking
Choose either Mol Table Ligand or Loaded Ligand
Interactive Docking - Mol Table Ligand If you have a chemical table already loaded into ICM you can use this option to dock them. You can read mol/mol2 or sdf files into ICM by using File/Open. They will be displayed in a table.
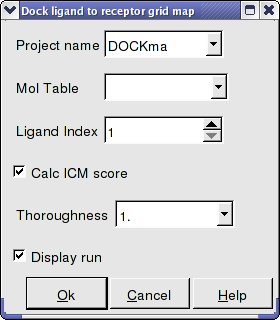
- Enter the Docking Project Name
- Use the drop down arrow to find the table of ligands you wish to dock.
- Enter the number of the ligand in the table you wish to dock. Eg if the ligand is in row 6 enter 6.
- If you have ICM-VLS you can retrieve an ICM docking score for the docked ligand.
- Thoroughness represents the length of the simulation. Generally 1 is a reasonable value for buried hydrophobic pockets. If you are docking to solvent exposed pockets or pockets containing metal ions you may wish to increase this slightly.
- Display run will display the ligand sampling the energy in the ligand binding pocket. Although this is fun to watch this significantly slows down the docking operation.
Interactive Docking - Loaded Ligand
If you have a ligand as an ICM object you can use this option.
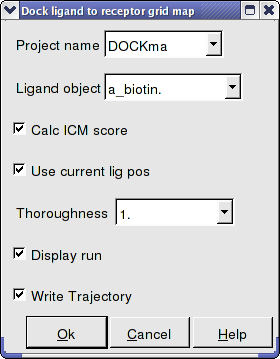
- Enter the Docking Project Name
- Use the drop down arrow to find the ligand.
- If you have ICM-VLS you can retrieve an ICM docking score for the docked ligand.
- If the ligand is already located in the pocket you can use this option. However by default the ligand will start sampling in the center of the pocket so this option does not need to be used.
- Thoroughness represents the length of the simulation. Generally 1 is a reasonable value for buried hydrophobic pockets. If you are docking to solvent exposed pockets or pockets containing metal ions you may wish to increase this slightly.
- Display run will display the ligand sampling the energy in the ligand binding pocket. Although this is fun to watch this significantly slows down the docking operation.
- You can write the docking simulation to a trajectory file. Please see the command language manual for more information on this.
17.1.10 Batch Docking |
Batch Docking is used for running docking jobs in the background. It is ideal for large-scale docking jobs.
- Docking/Batch Ligand Setup
- Select which format (see below) your ligand is in - object-loaded or file, mol/mol2, inx, MolCart.
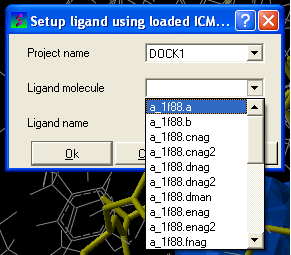
- Enter the name of the docking project followed by the ligand molecule name and you can also change the name of the ligand if you wish.
From File: ICM If your ligand (s) is saved and converted to an ICM object but is not yet loaded into ICM then you need to use this option.
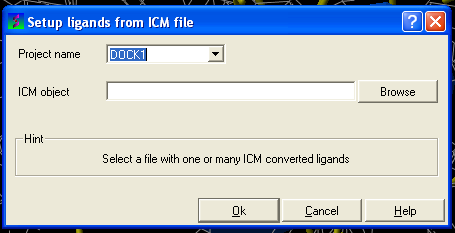
- Enter the name of the docking project.
- Click OK
From File:MOL/MOL2
If your ligand is a MOL or MOL2 file then
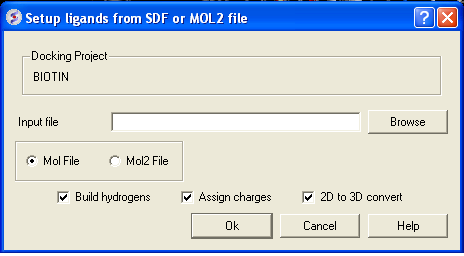
- Browse for your MOL/MOL2 file.
- Select whether your ligand is in MOL or MOL2 format.
- If you wish hydrogens to be added to your compound or charges to be assigned then click on the appropriate boxes in the display panel.
- Click OK
File Formats:
MOL Format
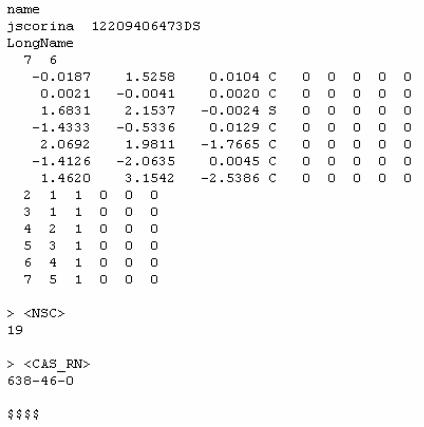
MOL2 Format

From Indexed Database - only available with ICM-VLS
In most cases the ligand input file will be an SDF or MOL2 file. These files need to be indexed by ICM before they can be used in VLS runs (see next section of this manual). The index is used to allow fast access to an arbitrary molecular record in a large file such as an SDF file which in some cases contains over one million compounds.
To index an sdf file:
- Click on the menu Docking/Tools/Index Mol/Mol2 File/Database to generate the index. The following data entry box will be displayed.
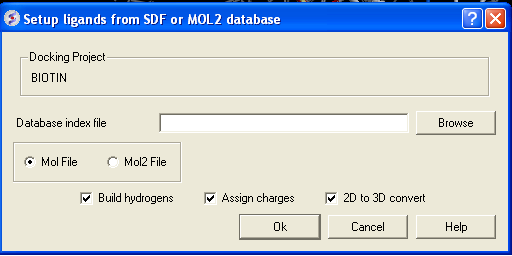
- Enter the name of your Mol/Mol2 file and enter the name you wish to call your index file.
- Select whether your file is in Mol or Mol2 format.
- Browse for your Index file.
- Select whether your ligand is in MOL or MOL2 format.
- If you wish hydrogens to be added to your compound or charges to be assigned then click on the appropriate boxes in the display panel.
- Click OK
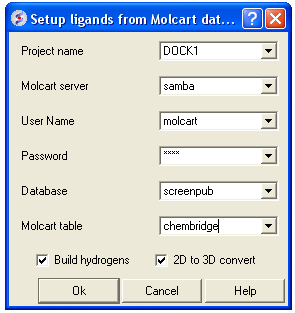
From MolCart - only available with ICM-VLS
| NOTE A separate license is required for MolCart |
- Enter docking project name.
- Enter the MolCart server
- Enter your username
- Enter Password
- Enter the Database Name
- Enter the name of the MolCart table within the Database
- Select whether you would like to build hydrogens or convert the compounds from 2D to 3D.
IMPORTANT - NOW SET YOUR BATCH JOB RUNNING USING DOCKING/SMALL SET DOCKING BATCH
| NOTE: NOW SET YOUR BATCH JOB RUNNING USING DOCKING/SMALL SET DOCKING BATCH} |
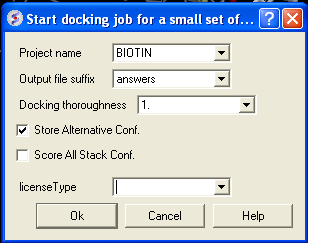
- Docking/ Run Docking Batch
- Enter the Docking Project Name
- Batch docking will generate an output file - enter the name you wish to call this file here.
- Thoroughness represents the length of the simulation. Generally 1 is a reasonable value for buried hydrophobic pockets. If you are docking to solvent exposed pockets or pockets containing metal ions you may wish to increase this slightly.
- Selecting Store Alternative Conf will allow you to look at all conformations in the energy stack.
- Score All Stack Conf. will allow you to determine an ICM docking Score for all members of the stack - this will slow the docking down.
- License type: For nearly all license (e.g. standard ICM-VLS licenses) types you need to leave this entry blank. If you have a -vlscluster license or a molvls license (-vls) then select the option from the drop down list. Any questions first check your license.dat file or Email support@molsoft.com.
- Click OK IMPORTANT - IF THE JOB IS RUNNING IT WILL TELL YOU bgrnd job AT THE TOP OF THE GUI - SEE BELOW
Docking simulation is running
![]()
Docking simulation has ended message
To check the status of your docking simulation
- Windows/Background Jobs
17.1.11 Viewing Your Docking Results |
Docking results can be visualized and browsed in one of the following ways.
- Docking/Browse/Scan Hits - If you have docked a multi-ligand file (eg SDF) or if you want to go back and see a single docked structure you can scan the structures using this option.
- Docking/Browse/Stack Conformation - View other possible conformations for the docked ligand ranked by energy.
- Docking/Make Hit List - Rank docked compounds by ICM Docking Score Only available with ICM-VLS
The results of the docking are saved in the following files
PROJECTNAME_LIGANDNAME.ob #icm-object file with best solutions for each ligand PROJECTNAME_*.cnf # icm conformational stack files with multiple docked conf.
The results of the docking job using ICM-VLS (separate license required) are saved in the folling files:
PROJECTNAME_answers*a.ob #icm-object file with best solutions for each ligand
PROJECTNAME_*.cnf # icm conformational stack files with multiple docked conf.
PROJECTNAME_*.ou # output file where various messages are stored eg.SCORE
17.1.12 Results - Scan Hits |
- Select Docking/Browse/Scan Hits and a data entry box as shown below will be displayed.
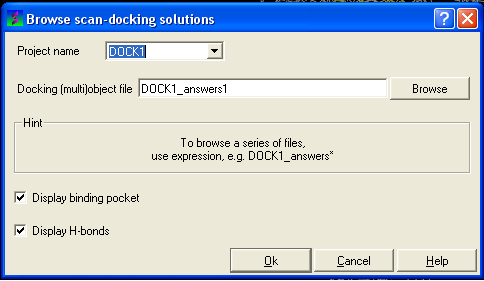
- Select the correct project name for the docking simulation results you wish to browse.
- Enter the name of the icm object file in the Docking (multi)object data entry field. This file will be called PROJECTNAME_answers*.ob or LIGAND_NAME.ob The browse button can be used to search for the correct file.
- You can display the binding pocket or the H-bonds by selecting the appropriate boxes in the Browse scan-solutions data entry window (shown above).
- Use the buttons at the bottom of the graphical user interface to browse the docked conformations. NEXT(or type �n�), BACK (or type �b�), JUMP (or type �j�), RETAIN (or type �r�), STOP (or type �s�), KEEP_STOP (or type �k�).
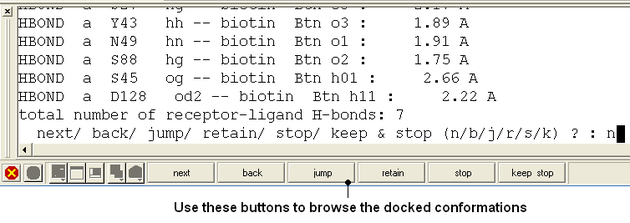
- The options keep and stop and retain will retain the displayed ligand in the graphical user interface. If you want to export the docked complex as a PDB file you will need to move the ligand and receptor into one ICM object. Moving objects is described in the FAQ section entitled How can I merge two objects into one?
17.1.13 Docking Results - View Stack Conformations |
To view the multiple positions of a single ligand in the docking simulation ranked by energy.
- Select menu Docking/Browse/Stack Conformations
The Browse Stack Conformation data entry window will be displayed.
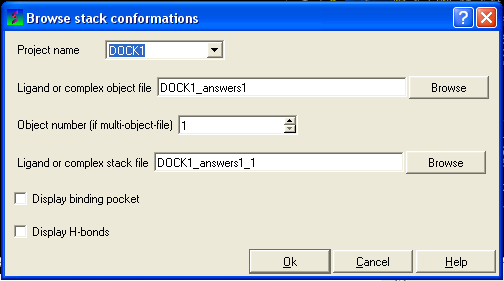
- Select the correct project name for the docking simulation results you wish to browse.
- Enter the name of the icm object file in the Docking (multi)object data entry field. This file will be called PROJECTNAME_answers*.ob .The browse button can be used to search for the correct file.
- Enter the name of the icm conformational stack files with multiple docked conformations into the Ligand or complex stack file data entry box. This file will be called PROJECTNAME1_1.cnf . The browse button can be used to search for the correct file. The second solution in the stack can be viewed by changing the number 1 at the end of the file name to 2 (PROJECTNAME1_2.cnf) and so on for each solution in the stack.
- You can display the binding pocket or the H-bonds by selecting the appropriate boxes in the Browse scan-solutions data entry window (shown above).
- Use the buttons at the bottom of the graphical user interface to browse the docked conformations. NEXT(or type �n�), BACK (or type �b�), JUMP (or type �j�), RETAIN (or type �r�), STOP (or type �s�), KEEP_STOP (or type �k�).
- The options keep and stop and retain will retain the displayed ligand in the graphical user interface. If you want to export the docked complex as a PDB file you will need to move the ligand and receptor into one ICM object. Moving objects is described in the FAQ section entitled How can I merge two objects into one?
Columns in the Stack Table
i rank in stack
ener Energy kcal/mol
gvw van der Waals grid potential
gb hydrogen bonding grid potential
ge electrostatic grid potential
gs hydrophobic grid potential
Einternal is internal conformation energy of the ligand
17.1.14 Make a HIT LIST - Only available with ICM-VLS |
- Docking/Make Hit List
- Enter project name
- Use the browse button to locate DockingProjectName_answers.ob file
- You can include a 2D image into the HITLIST
- Select Unique if you have made multiple docking runs the best docking score will be taken to make the hitlist unique.
- A HITLIST table will be displayed. Each docked ligand can be viewed by double clicking in the HITLIST table. A stack of conformations for each ligand will also be displayed in a table.
Columns in the HitList Table
IX is the index number from the docked database
Score is the ICM score -32 and lower are generally considered good scores - but depends on the receptor (e.g. exposed pockets or pockets with metal ions mayhave higher scores than -32).
Natom is the number of atoms in docked ligand
Nflex is the number of rotatable torsions.
Hbond is Hydrogen Bond energy
Hphob is the hydrophobic energy in exposing a surface to water
VwInt is the van der Waals interaction energy (sum of gc and gh van der waals). Current version of the score uses explicit van der Waals interaction energy calculation (no grids)
Eintl is internal conformation energy of the ligand
Dsolv is the desolvation of exposed h-bond donors and acceptors.
SolEl is the solvation electrostatics energy change upon binding.
mfScore is the potential of mean force score
RecConf - if multiple receptor conformations was used Docking/Flexible Receptor/Setup 4D grid and represents the receptor conformation number.
17.1.15 Reload a Docking Project |
To reload a docking project.
/Docking/Set Project - Type in the Docking Project Name (Case Sensitive)
Now you can browse scan solutions etc.... and use the maps to dock another ligand.
| Prev Docking | Home Up | Next Flexible Docking |