June 6th 2026 - MolSoft and XtalPi Partner to Unlock the 4.66 Billion Compound VAST™ Space for 3D Docking
San Diego, CA — June 6, 2026 — MolSoft LLC is excited to announce a new partnership with XtalPi, bringing the power of the MolSoft ICM computational platform directly to XtalPi’s massive VAST™ chemical universe.
This collaboration enables researchers to perform ultra-large-scale virtual 3D screening against XtalPi’s newly released VAST™ Space, a virtual library containing 4.66 billion synthetically accessible, drug-like molecules designed for rapid hit discovery and lead generation.
Efficient exploration of multi-billion compound libraries depends on highly optimized docking workflows capable of balancing throughput, accuracy, chemical diversity, and synthetic accessibility. MolSoft’s CombiRIDGE platform provides the infrastructure needed to meet this challenge through a combination of GPU acceleration, AI-enhanced enumeration, and physics-based molecular docking.
“Accurately exploring a chemical space of this magnitude requires a fundamental shift in how we approach ligand 3D docking and binding energy evaluation,” said Ruben Abagyan, Ph.D., Founder of MolSoft. “We have developed rigorous, physics-based conformational sampling and flexible receptor docking to prioritize true binders. Combining our CombiRIDGE infrastructure with XtalPi's highly curated, synthesis-aware space means our users can confidently discover novel chemotypes that are biologically active and instantly accessible. The CombiRIDGE approach has already had impressive hit-rates screening other ultra-large libraries for our Pharmaceutical and Biotech partners.”
— Ruben Abagyan Ph.D., MolSoft Founder
The MolSoft ICM platform has been extensively validated across difficult targets and large-scale industrial screening campaigns. Using advanced scoring methodologies, flexible docking, and efficient conformational sampling, CombiRIDGE enables practical screening of ultra-large chemical spaces while maintaining the rigorous accuracy required for structure-based drug discovery.
XtalPi’s VAST™ Space is uniquely architected upon more than 40 rigorously validated chemical reaction protocols (spanning complex cross-couplings, macrocyclizations, and precise functional group transformations) and over 58,000 carefully curated, commercially available building blocks. The platform utilizes advanced graph neural networks (GNNs) and automated retrosynthetic planning to evaluate chemical feasibility, achieving an expected synthetic success rate greater than 85%. Crucially, these molecules can typically be realized within one to two synthetic steps, drastically reducing the attrition rate historically associated with massive virtual libraries.
“By combining MolSoft’s high-performance virtual screening technology with XtalPi’s scalable synthesis-ready chemical space, the partnership creates a streamlined workflow from computational hit identification to rapid compound realization.” A virtual hit is only valuable if it can be rapidly synthesized and tested on the bench. By embedding our VAST™ Space, powered by robust AI-driven synthetic feasibility predictions, directly into MolSoft's highly accurate ICM ecosystem, we are closing the loop. Researchers can now transition computationally identified hits into experimentally testable compounds in a matter of weeks, dramatically accelerating the hit-to-lead lifecycle.”
— Greg Pickler, Senior Director of Business Development at XtalPi
The integration of VAST™ Space into the MolSoft ICM ecosystem provides researchers with direct access to billions of readily synthesizable molecules for docking, hit expansion, scaffold hopping, and lead optimization campaigns across a wide range of therapeutic targets.
Together, MolSoft and XtalPi are enabling a new generation of ultra-large-scale, synthesis-aware virtual screening workflows designed to accelerate modern drug discovery.
About MolSoft LLC
MolSoft is a premier developer of leading-edge software tools for computational biology and chemistry. Utilizing its proprietary Internal Coordinate Mechanics (ICM) methodology, MolSoft offers highly accurate solutions for molecular docking, structure-based drug design, cheminformatics, and protein modeling. MolSoft's platforms are utilized by top pharmaceutical and biotechnology companies and academic institutions worldwide.
About XtalPi
XtalPi is a pioneering pharmaceutical technology company fundamentally transforming drug discovery and development through its quantum physics-based, AI-driven computation and robotic automation platform. With a mission to discover highly effective therapies for patients globally, XtalPi seamlessly integrates cutting-edge computational chemistry, machine learning, and automated wet-lab synthesis to accelerate the delivery of novel therapeutics.
May 20th 2026 - ICM-Pro Drug Discovery Workshop in Seoul
Gangnam, Seoul, South Korea — May 23, 2026 — MolSoft LLC is pleased to announce the completion of an intensive, structural biology and computer-aided drug design (CADD) workshop hosted by our collaborative partners, inCerebro Co., Ltd. The session, held on May 20th in Gangnam, Seoul, provided local researchers with advanced hands-on training using MolSoft’s flagship ICM-Pro software platform.
The technical curriculum focused on deploying ICM's global optimization algorithms and AI methods to solve problems in structure-based drug discovery. Participating research teams from regional pharmaceutical corporations and academic laboratories explored advanced molecular modeling workflows, mapping protein-ligand interaction networks with high atomic precision.
Key technical modules included:
Advanced Target Preparation: Optimizing protein structures, resolving ambiguous electron density features, and sampling alternative side-chain conformations.
Allosteric & Cryptic Pocket Identification: Utilizing the icmPocketFinder grid-based mapping algorithm to locate and analyze potentially targetable, interface-adjacent binding sites.
High-Throughput Virtual Ligand Screening (VLS): Setting up robust biased docking screens using internal coordinate mechanics, allowing for the precise conformational sampling of large, chemically diverse compound libraries.
The session demonstrated how researchers can maximize computational throughput and utilize parallel hardware setups to accelerate the hit-to-lead optimization process. The technical value of the platform resonated strongly with the attendees, generating immediate follow-up inquiries regarding the deployment of specialized ICM-Pro configurations within active discovery pipelines.
MolSoft extends its gratitude to the scientific team at inCerebro for organizing and delivering this exceptional session—specifically presenters Minkyu Kim, Chaeyoung, and Yesul—and to Art Cho for a highly successful and expanding research collaboration.
April 30th 2026 - MolSoft ICM and 3D Ligand Editor Power Discovery of Next-Generation WEE1 Kinase Inhibitors for Colorectal Cancer
San Diego, CA — A new study published in the European Journal of Medicinal Chemistry by Syphers et al. from Professor Jonathan Baell’s research group and collaborators details the discovery of a highly potent and selective class of WEE1 kinase inhibitors. The breakthrough research, aimed at treating aggressive colorectal cancers, heavily relied on MolSoft LLC’s Internal Coordinate Mechanics (ICM) software suite and its interactive 3D Ligand Editor to guide structure-based design.
The study highlights the identification of APO-50815, a leading next-generation compound that exhibits exceptional therapeutic efficacy. When tested in TP53-mutated colorectal cancer (CRC) patient-derived organoid (PDO) models, APO-50815 demonstrated outstanding anticancer activity, outperforming notable clinical trial comparators such as AZD1775 (adavosertib) and ZN-c3.
Driving Lead Optimization via 3D Structure-Based Design
To bypass costly, trial-and-error chemical synthesis, the research team implemented a rigid structure-based design strategy powered directly by MolSoft ICM. Utilizing the 3D Ligand Editor, the investigators were able to rapidly evaluate molecular binding hypotheses, visualize atomic interactions in real time, and optimize the molecular scaffold with precision.
The physics-based modeling workflow proceeded through several key stages:
System Preparation: The team initiated modeling from the co-crystal structure of WEE1 kinase bound to AZD1775 (PDB code: 5V5Y), carefully preparing the protein-ligand system at physiological pH to accurately simulate biological conditions.
Scaffold Modification: Using the interactive 3D Ligand Editor, they replaced the N-methyl piperazine tail moiety of AZD1775 with a tetrahydroisoquinoline (THIQ) group.
Redocking and Refinement: Real-time redocking simulations within ICM identified that the newly added THIQ ammonium group formed a critical, highly favorable salt bridge interaction with the side chain of Aspartate 386 (D386) in the WEE1 binding pocket.
Conformational Stabilization: Additional structural stabilization of the leading compound was rationalized and validated via an intramolecular cation–π interaction with the adjacent pyridyl ring.
A Validated Approach for Modern Medicinal Chemistry
The success of APO-50815 highlights the immense value of tightly integrating interactive 3D ligand optimization with robust, physics-based and AI scoring functions. By allowing medicinal chemists to directly manipulate structures within the digital binding pocket and receive instantaneous feedback on binding affinity, MolSoft ICM continues to streamline the transition from initial hit to optimized clinical candidate.
January 12th 2026 - ICM-Pro v3.9-5 Released: New AI and Physics-Based Capabilities for Drug Discovery
MolSoft is pleased to announce the release of ICM-Pro v3.9-5, a major update that introduces new capabilities for structure-based drug discovery, lead optimization, and ultra-large chemical space exploration.
Version 3.9-5 is now available from the MolSoft support site. A summary of the most significant new features is provided below. For additional details, see the new features page.
CombiRIDGE is a new GPU-accelerated workflow for efficient screening of ultra-large and combinatorial libraries. Starting from a pre-docked 3D anchor and associated 2D R-groups, CombiRIDGE generates full 3D conformers directly in the binding site using a neural network model. The resulting poses are refined with GPU acceleration and evaluated using RTCNN scoring, enabling rapid R-group optimization while preserving key binding interactions.
groupGen – Generative AI for Lead Optimization
groupGen is a context-aware generative neural network that proposes R-group substitutions tailored to a specific protein binding site. Using a 3D graph neural network representation of the local receptor environment, groupGen generates chemically realistic and sterically compatible substituents. Generated analogs are automatically docked with CombiRIDGE and prioritized using RTCNN, VLS scores, and synthesizability metrics.
Ligand AIDE – De Novo Ligand Design by AI-Driven Evolution
Ligand AIDE introduces an iterative evolutionary workflow for de novo ligand generation. Starting from docked fragments, compounds are grown and optimized over multiple generations using generative substitutions, re-docking, and selection based on binding scores, drug-like properties, and synthesizability.
Comprehensive Peptide Modeling in the 3D Interactive Ligand Editor
ICM-Pro v3.9-5 adds full peptide modeling capabilities to the Interactive Ligand Editor. New functionality includes residue-level editing, continuous-chain handling, peptide–receptor co-minimization, RTCNN scoring, secondary structure refinement, and support for stapled peptide modeling.
GigaSearch v2.0 – Giga-Scale Chemical Searching
GigaSearch v2.0 enables interactive similarity and substructure searching across libraries containing billions to hundreds of billions of compounds. Optimized fingerprints and statistical pruning allow queries to complete in seconds, with direct integration into ICM-Pro workflows.
Neural Network–Based pKa Prediction Updates
Updated ensemble neural network models improve pKa prediction accuracy, tautomer ranking, and protonation state assignment. A new GPU-accelerated workflow enables fast and consistent formal charge determination for large compound sets.
Additional Improvements
Binary MPO classification using Random Forest models
New training and test set splitting tools, including Kennard–Stone methods for diverse dataset selection
2025
November 12th 2025 - MolSoft Releases GigaSearch v2.0 - Efficient Massive Scale Substructure and Similarity Chemical Searching
San Diego, CA — November 12th 2025 MolSoft LLC announces GigaSearch v2.0, a next-generation chemical search engine capable of scanning billions to trillions of compounds within seconds. Built for ultra-large substructure, similarity and analog searches, GigaSearch v2.0 enables rapid exploration of vast chemical spaces such as Enamine REAL, Chemspace Freedom, and other make-on-demand libraries.
Key Features of GigaSearch v2.0
Speed: The average search time is typically around 5 to 10 seconds on the 80B Enamine Space and 140B Chemspace Freedom database.
Storage Efficiency: GigaSearch v2.0 handles much larger datasets with vastly less storage e.g., ~86 GB for the 80B 2025 Enamine Space.
Support for SMILES and SMARTS Search: Advanced substructure queries allow chemical patterns with atomic and bonding properties, including atom type, aromaticity, ring membership, hybridization, valence, charge, and connectivity.
Scalability: Fully utilizes multicore systems for rapid, large-scale searching.
GigaSearch v2.0 can be accessed directly via the MolSoft server through the Chemical Search → Online Databases tab in ICM-Pro and ICM-Chemist, or installed locally under license for in-house high-speed screening.
October 1st 2025 - MolSoft Unveils CombiRIDGE: Next-Generation GPU-Accelerated Virtual Screening for Ultra-Large Chemical Spaces
San Diego, CA — October 1st 2025 — MolSoft LLC today announced the launch of CombiRIDGE, a GPU-accelerated combinatorial docking and virtual screening solution designed to efficiently explore ultra-large chemical spaces. CombiRIDGE combines neural networks, fast GPU docking, and modern scoring functions to accelerate hit discovery and lead optimization.
A New Paradigm for Combinatorial Docking
CombiRIDGE enables drug hunters to optimize and evaluate R-groups around a defined core fragment, referred to as an anchor, to rapidly generate and score complete molecules directly in a protein binding site. Rather than enumerating full libraries in advance, CombiRIDGE grows combinatorial libraries on-the-fly, allowing efficient navigation of extremely large chemical spaces.
Core Technologies Behind CombiRIDGE
GINGER is MolSoft’s neural network-based conformer generation technology. It generates accurate three-dimensional conformations of anchor fragments directly in the context of the protein binding site, ensuring that downstream docking starts from realistic ligand geometries.
RIDGE is MolSoft’s GPU-optimized docking engine. It enables extremely fast docking of millions of anchor conformations, making it possible to screen very large libraries using a small number of modern GPUs.
RTCNN is a graph neural network-based scoring function trained to evaluate protein-ligand interactions. RTCNN provides robust ranking of docking poses and helps prioritize high-quality candidates for further expansion and experimental follow-up.
September 17th 2025 - MolSoft and NIH Collaboration: Novel GPU Engines Conquer Challenging Targets
Molsoft scientists have successfully developed and deployed powerful GPU-accelerated virtual screening engines to rapidly screen chemical libraries containing billions of compounds. In collaboration with Nadya Tarasova's group at the NIH this technology has led to the discovery of potent drug leads against two historically "undruggable" cancer targets, K-Ras G12D and PD-L1. Read the paper here.
The ability to screen massive chemical spaces is vital for next-generation drug discovery. Molsoft's GPU implementations achieve this with unprecedented speed:
RIDGE (Rapid Docking GPU Engine): A novel structure-based docking method running on the GPU. RIDGE provides an approximately 10-fold speed boost over previous state-of-the-art GPU docking, processing ~100 compounds per second per GPU.
RIDE (Rapid Isostere Discovery Engine): A powerful GPU-accelerated ligand-based 3D similarity screening method. Its parallel processing allows it to search approximately 0.5 million conformers per second per GPU, making giga-scale screening efficient.
The paper describes the discovery of potent hits against two difficult cancer proteins.
Programmed Death-Ligand 1 (PD-L1): The screens identified five novel small-molecule inhibitors of the PD-L1 immune checkpoint. One compound, identified by RIDE, demonstrated high potency with an IC
50 of 0.19 uM.
K-Ras G12D: Against this highly challenging mutation, RIDE successfully identified three novel binders from billions of compounds. One compound, 7rpz-2, caused a substantial melting temperature shift
in experimental validation, confirming potent binding and providing an excellent starting point for optimization.
The publication reports novel GPU-accelerated screening methods that significantly outperform conventional virtual ligand screening (VLS). MolSoft's docking scores proved to be a more accurate predictor of binding, leading to the successful identification of eight potent inhibitors (five for PD-L1, three for K-Ras G12D) with submicromolar affinities. These powerful new methods effectively expand the range of molecules MolSoft can screen and enable hit identification even for challenging drug targets.
March 7th 2025 - MolSoft Release a White Paper on the RTCNN Score which Outperforms other Scoring Methods for Virtual Screening
MolSoft today released a white paper about the RTCNN - Radial and Topological Convolutional Neural Network Score which is outperforming other Virtual Screening scoring methods. The RTCNN score is used in all MolSoft's structure-based screening methods including ICM-VLS, RIDGE, GigaScreen and CombiRIDGE.
Ligand scoring plays a crucial role in virtual screening, a computational technique used to identify potential drug candidates from large compound libraries.
Graph Convolutional Neural Networks (GCNNs) have shown promise in improving ligand scoring by capturing the structural and chemical features of molecules in a graph representation. Traditional ligand scoring methods rely on handcrafted features and simplified physical energy terms that may not fully capture the complex interactions between a ligand and its target protein. GCNNs offer a data-driven approach by learning directly from the molecular graph representation, allowing for more accurate and robust scoring.
February 21st 2025 - NIH Publish PROTAC Modeling Success using MolSoft ICM
A new study published today in RSC Medicinal Chemistry by Dr. Burke's lab at the NIH National Cancer Institute (NCI) explores the development of proteolysis-targeting chimeras (PROTACs) to selectively degrade tyrosyl-DNA phosphodiesterase 1 (TDP1). The paper, titled "Application of a Bivalent "Click" Approach to Target Tyrosyl-DNA Phosphodiesterase 1 (TDP1)," highlights the potential for PROTACs to enhance the efficacy of topoisomerase 1 (TOP1) inhibitors used in cancer therapy.
Using MolSoft ICM, the researchers modeled the ternary structure of a PROTAC complex, docking a lead imidazopyridine-based TDP1 inhibitor (compound 1b) with thalidomide to form a bivalent conjugate (compound 5h). The modeling results confirmed that the linker in 5h was optimally positioned, extending from the peptide-binding groove of TDP1 toward the CRBN binding site, supporting the intended PROTAC mechanism.
This study provides valuable insights into the structure-based design of TDP1-targeting PROTACs, paving the way for future therapeutic advancements.
December 6th 2024 - Advent Informatics Host Successful ICM User Group Meeting in India
Advent Informatics, the distributor of MolSoft's ICM-Pro software in India, recently hosted a well-attended and engaging User Group Meeting. The event featured compelling success stories in areas such as small-molecule drug design and discovery, PROTAC development, and antibody design. It also showcased innovative approaches for ultra-large library screening, multiple receptor docking, and covalent docking. The meeting included insightful presentations by scientists from leading pharmaceutical and biotech companies. The following speakers presented their research:
Leads from Space: Screening billions for a preclinical candidate - Dr. Ruben Abagyan, Molsoft LLC
Driving Innovation in Virtual Screening: High Accuracy and Evolving Methods with ICM-Pro from Targeted Libraries to Ultra-Large Scale - Dr. Andrew Orry, Molsoft LLC
Modelling Bifunctional Protein Degraders: Some in-house Success Stories - Dr. Subhendu Mukherjee, Aurigene Oncology Limited
4D Docking and Other Tools: Scaling the ICM learning curve - Dr. Ramesh Sistla, Think Molecular Technologies Pvt Ltd
Success Stories in Small Molecule and Antibody Design: Dr. Tripuraneni Srinivas, Syngene International Ltd.
Covalent Docking in ICM: Transforming Drug Discovery with Precision and Innovation - Dr. Pathan Mohsin Khan, Advent Informatics Pvt. Ltd.
Modelling and Rapid Synthesis of RIPK-2 Degraders: Improved ADME and PK Parameters using in-house partial PROTAC library - Dr. Sunil Kumar Panigrahi, Aurigene Pharmaceutical Services Ltd.
A gallery of photos from the event can be viewed on Advent Informatic's website here.
September 17th 2024 - ChemSpace Scientists Achieve Success in the CACHE Challenge by Screening Billions of Chemicals using V-SYNTHES MolSoft ICM-Pro
MolSoft LLC is pleased to share that ChemSpace has completed the CACHE 2 Challenge with notable success, utilizing MolSoft's ICM software suite. Leveraging MolSoft's ICM tools, Chemspace achieved impressive results in high-throughput virtual screening, significantly advancing the discovery of promising drug candidates for the COVID target NSP13 helicase.
The results highlights the capabilities of MolSoft's ICM platform, which enabled Chemspace to screen large ligand libraries (176B chemicals) accurately and at high speed. This outcome demonstrates the robustness of MolSoft's technology in screening ultra-large libraries, enabling efficient and accurate identification of high-quality hits Read more on ChemSpace's website here.
April 26th 2024 - New Publication on Molsoft's GINGER method for lightning-fast high quality conformer library generation on GPUs.
A paper by MolSoft's scientists and developers Eugene Raush (Princpal Developer), Ruben Abagyan (MolSoft Founder) and Maxim Totrov (CTO) has today been published in the ACS Journal of Chemical Theory and Computation. The paper unveils a novel cutting-edge algorithm called GINGER, which efficiently generates high quality chemical conformers at lightning-fast speeds. This innovative approach leverages neural networks to rapidly predict low-energy conformers from 2D chemical structures. GINGER's efficiency is further enhanced by GPU optimization, enabling rapid processing of large compound libraries.
GINGER can convert 150-200 mol/sec on a single Nvidia RTX 4090 GPU making it suitable for the efficient conversion of ultra-large libraries (>1B molecules) when run on a multi-GPU server. The ultra-large libraries can be screened using MolSoft's rapid field-based 3D similarity search engine RIDE or via fast structure-based docking use RIDGE and GigaScreen.
GINGER is a separate add-on module to MolSoft's desktop modeling software ICM-Pro and ICM-Chemist-Pro or can be run by MolSoft as a service. Please contact MolSoft with any questions and to obtain an evaluation license or to purchase GINGER.
April 19th 2024 - MolSoft ICM Drives GPU-Enhanced Structure- and Ligand-Based Screening of LCC's Novel Ultra-Large, Stereo-defined, and Diverse Library
LCC's ultra-large library has been converted to 3D conformers using Molsoft's new highly accurate neural network GINGER conformer generator. The database is based on LCC's proprietary collection of chirally pure, sp3-rich scaffolds with a diverse set of ring sizes, ring topologies, and exit vectors. The reaction-based enumeration facilitates transformation of your chiral virtual hits to high-quality physical samples.
MolSoft offers a cost-effective service for rapidly screening the database, with results analyzed by our team of computational chemistry experts. Virtual hits then can be converted to physical compounds using LCC's efficient parallel synthesis laboratories. The library can be screened by MolSoft either through a structure- or ligand-based approach as a service.
Structure-Based Lead Discovery
If atomic resolution structures or AlphaFold models of your target protein or nucleic acid are available, screening the LCC database against the structure can be undertaken using MolSoft's high-performance GPU-based RIDGE-docking or deep learning GigaScreen methods. The top scoring compounds will undergo inspection, and a ranked hitlist will be provided along with 3D poses of the top hits in complex with the receptor along with other essential properties.
Ligand-Based Isostere Scaffold Hopping Discovery
If one or more lead chemicals are available, the library can undergo screening using MolSoft's Rapid Isostere Discovery Engine (RIDE) approach. This will enable new chemistry with similar 3D pharmacophore properties and shape to be identified from the LCC database. Atom weighting can be employed to highlight the significance of specific moieties, while excluded volumes penalties can be applied to regions surrounding the query molecule, prioritizing hits without bulky extensions in constrained areas.
MolSoft's screening methods are finely tuned for high-performance GPUs, allowing us to swiftly deliver screening results to your company within 1-2 weeks in most cases.
Please contact MolSoft if you have any questions or wish to schedule a call to discuss your project and MoSoft's virtual screening approach of the LCC database. Please contact LCC if you have any questions regarding the ultra-large chemical library.
About Molsoft
Molsoft LLC is a leading provider of Physics- and AI- based scientific software solutions, specializing in computational chemistry, biology, and molecular modeling. MolSoft's flagship product, ICM (Internal Coordinate Mechanics), equips researchers in academia, pharmaceuticals, and biotechnology with powerful tools for molecular design, virtual screening, and drug discovery. With a dedication to in silico method innovation, Molsoft is committed to advancing the frontiers of scientific research and pharmaceutical development.
About Liverpool ChiroChem
LCC is an established, chemical technology innovator, on a mission to accelerate the discovery and development of high-quality drugs. LCC provide expanded access to 3D chemical space to support orthogonal hit-ID (DEL, FBLD, VLS), hit optimisation using our parallel synthesis laboratory, and personalised support using our design and computational team to expedite targeted chemical space exploration.
March 30th 2024 - MolSoft Founder will Chair AI/ML Drug Discovery Session and Present at Upcoming Drug Discovery Chemistry Conference in San Diego Apr1-4 2024
MolSoft's founder Ruben Abagyan Ph.D. and Prof at UCSD will be chairing a session at the Drug Discovery Chemistry Conference in San Diego on April 4th 2024. The title of his
talk is "AI-ML Docking Pipeline for Giga-Screens versus New Target Profiles and Hidden Pockets" and he will be chairing the AI/ML for Target-Specific Applications session.
2023
December 19th 2023 - Best in Class BCL2 and FLT3 Inhibitors Reported at ASH 2023
A collaboration between MolSoft, Eilean Therapeutics, Expert Systems, ChemDiv, and John Byrd's laboratory at the University of Cincinnati reports some exciting discoveries at the 66th Annual American Society of Hematology meeting 2023.
Please see the links to the posters describing new best in class BCL2 and FLT3 inhibitors below:
A novel Selective BCL2 inhibitor with limited immune suppression and improved safety compared to venetoclax at the American Society of Hematology Annual Meeting. You can view the poster here.
Therapeutic Targeting of FLT3 gate keeper mutation with E2082-0047 in traditional and a novel Immunocompetent murine adoptive transfer model of AML. You can view the poster here.
November 30th 2023 - MolSoft Releases GINGER - Graph Internal-coordinate Neural-network conformer Generator with Energy Refinement for Large Chemical Libraries
Conformer generation is an essential step of a variety of molecular modeling and computer-assisted drug discovery workflows such as 3D ligand-based virtual screening or fast GPU docking. GINGER (Graph Internal-coordinate Neural-network conformer Generator with Energy Refinement) is Molsoft's new cutting-edge software designed for lightning-fast high quality conformer library generation on GPUs.
October 5th 2023 - MolSoft ICM-Pro Now available on latest MacOS Sonoma
Good news for users of ICM on a Mac. ICM-Pro is fully compatible with the latest MacOS Sonoma - get in contact with us to obtain the MacOS Sonoma package.
August 24th 2023 - Eilean Therapeutics Doses the First Patient with ZE46-0134, a selective FLT3wt-SPARING, PAN FLT3mut Inhibitor
Eilean Therapeutics LLC is a company that utilizes MolSoft's AI and Physics-Based molecular modeling tools to rationally develop first and best in class therapeutics.
You can read Eilean Therapeutic's recent press release below:
Eilean Therapeutics LLC, a biopharmaceutical company dedicated to discovering and developing best-in-class and first-in-class small molecule inhibitors to target escape mutations in hematologic malignancies, today announced the initiation of human dosing in the Phase 1 clinical program of ZE46-0134, a highly potent and selective pan-FLT3 inhibitor that targets clinically relevant FLT3 mutations including the FLT3 gatekeeper mutation, while sparing the wild type form of the protein. Read More...
May 18th 2023 - Successful Exciting ICM User Group Meeting - Watch the Highlights
On May 17-18, 2023 MolSoft hosted its highly anticipated User Group Meeting in an online format. The event featured engaging talks by both our esteemed users and talented developers, spotlighting the exciting array of features present in ICM. Attendees learnt about the latest cutting-edge technologies, including GPU-based and AI-accelerated virtual screening, structure- and ligand-based drug design, PROTAC modeling, membrane protein modeling, and much more.
You can watch a selection of the presentations again on our YouTube channel - see the links below:
New technologies in Molsoft ICM: neural networks and beyond. Maxim Totrov PhD (CTO - MolSoft) YouTube
Highlights of recent ICM developments: GPU acceleration, its applications and more... Eugene Raush (Principal Developer, MolSoft) YouTube
Mining in the Ultra-Large Library SAVI with the Help of Molsoft Tools. Marc Nicklaus Ph.D. (NCI). YouTube
Don't blame the software. It is the structure, bro. Nadya Tarasova Ph.D. (NCI). YouTube
How Molsoft Scarab helped fight COVID-19 in the UK - Brian Marsden Ph.D. University of Oxford. YouTube
Computer Driven Discovery of GPCR Ligands with New Chemotypes and Functional Selectivity Vsevolod "Seva" Katritch Ph.D. (USC). YouTube
Computational approaches and methodologies in proximity induced pharmacology. Evianne Rovers (SGC, University of Toronto). YouTube
Design and Development of iPAI molecules for quantitative assessment of cancer drug target availability. William Bisson Ph.D. (OHSU Knight Cancer Institute). YouTube
Chemically probing the mechanism of KATP channels Tim Cardozo M.D. Ph.D. (NYU) YouTube
April 12th 2023 - MolSoft's Founder Prof. Ruben Abagyan Presents and Chairs Panel at Artificial Intelligence for Early Drug Discovery Conference.
Prof. Ruben Abagyan (UCSD, MolSoft Founder) presented at and chaired the panel for the session "Artificial Intelligence for Early Drug Discovery" at the Drug Discovery Chemistry Conference in San Diego this week.
The title of his presentation was "Giga-Screening for Preclinical Candidates with a Defined Multi-Target Profile" and his talk discussed the challenges involved with drug discovery of "tough targets" such as FLT3 and BCL2. Rational design of first- and best- in class candidates was achieved under two years using MolSoft's AI and Physics Based molecular modeling tools. The candidates for primary and drug resistant leukemias and lymphomas are initiating clinical studies. You can read more about this work at i2020 Accelerator here.
2022
December 3rd 2022 - New MolSoft ICM Paper - Graph-Convolutional Neural Net Model of the Statistical Torsion Profiles for Small Organic Molecules.
A new MolSoft ICM paper has been published in the J.Chem.Inf.Model - you can read it here. The paper describes a new graph-convolutional neural network (GCNN)-based method which can be used for learning and prediction of statistical torsional profiles (STP). The model has been trained on the Crystallography Open Database and has good correlation with quantum chemically calculated profiles. The method can be used for conformer generation and you can try the method here.
November 10th 2022 - New ICM Version 3.9-3 Released
MolSoft is excited to announce the release of a new ICM-Pro version 3.9-3. The new version contains a Torsion Profile Neural Network (NN) Prediciton Engine, Protonation State of Ligand vs pH tool, Compact Efficient GIGA Sized Screening Libraries, Automated PROTAC Modeling, Molecular Dynamics (MD), New NN Ligand Docking and Screening Score and much more...
Click here for a summary of the new tools and features and links to documentation.
Subscribe to MolSoft's YouTube Channel to keep up to date with latest training videos.
March 28th 2022 - Efficiently Screen Billions of Chemicals in MolSoft's ICM Software - A new collaboration between MolSoft, Enamine and the NIH
At MolSoft we have been privileged to collaborate with many exceptionally talented Ukrainian scientists and we aim to preserve those ties during this tragic time in Ukraine. Ukrainian companies such as Enamine and ChemSpace are currently relocating to other countries so as to keep serving their customers. We believe that one way the MolSoft ICM community can support them during this time is by maintaining the use of their products and services as normal, and providing them with our services.
You can now use MolSoft's Rapid Isostere Discovery Engine ( RIDE) method to efficiently screen over 48 billion chemical conformers from the Enamine REAL database. This will allow you to find new leads and scaffold hop to new interesting chemistry to aid your drug discovery program and quickly find new drugs for many kinds of diseases. MolSoft has collaborated with the NIH and Enamine to prepare Enamine's REAL database for virtual screening . The REAL database is a reaction enumerated library containing approximately 2 billion chemicals (read more here).
Ruben Abagyan Ph.D. (MolSoft Founder) said "We are excited to collaborate with Enamine and the NIH on another Giga sized screening project. RIDE is already being used routinely by our customers to screen the Giga-sized NIH SAVI library and we have worked with Enamine in the past to develop an efficient search method for REAL. We are happy that our customers will have another option for their drug discovery needs."
Nadya Tarasova Ph.D. (NIH/NCI) said "It was a big scientific and computation challenge to prepare the REAL library conformer database and it took more than 7 million CPU hours to complete but now the library is ready we are excited to see how it will be used by the scientific community to deliver new drugs to the market. We have already started screening the REAL database using MolSoft's RIDE method for drug discovery projects here at the NIH."
Vladimir Ivanov Ph.D. (Chief Sales and Marketing Officer at Enamine) said "We are happy that MolSoft's ICM user community will have access to efficiently screen the REAL database using the RIDE method. At Enamine we have enabled a high synthesis success rate for the REAL molecules and can make them available within 3-4 weeks. We look forward to working with MolSoft and the ICM users to help provide chemicals for their drug discovery projects."
MolSoft's RIDE is a fast 3D molecular similarity search method based on Atomic Property Fields (APF) invented by Molsoft Lead Scientist, Max Totrov, and implemented by Molsoft Lead Software Engineer Eugene Raush and his team. RIDE searches databases of compound conformers for molecules that are isosteric to the query, i.e. have similar 3D configurations and distributions of atomic properties. RIDE is a multi-conformational optimal 3D superposition in which both the query molecule and the molecule in the database are represented in multiple low energy conformations to reflect the flexible nature of both molecules. RIDE is available in the ICM-Pro + VLS package.
Applications screening the REAL database with RIDE include:
Scaffold hopping - discover structurally novel chemicals based on a lead.
Hit follow-up - quickly discover new chemicals with similar 3D pharmacophoric binding properties to your lead.
Core replacement in the ICM 3D ligand editor.
If you want to learn more about RIDE please see a recorded webinar here on YouTube.
The REAL database is saved in MolSoft's compact MOLT file format - please contact MolSoft for download instructions. Other RIDE formatted databases, including the NIH Giga Sized SAVI database are available here.
MolSoft Statement in Support of Ukrainian Chemists
At MolSoft we have been privileged to collaborate with many exceptionally talented Ukrainian scientists and we aim to preserve those ties during this tragic time in Ukraine. Ukrainian companies such as Enamine and ChemSpace are currently relocating to other countries so as to keep serving their customers. We believe that one way the MolSoft ICM community can support them during this time is by maintaining the use of their products and services as normal, and providing them with our services.
2021
MolSoft's ICM provides the Engine for 11 Billion Chemical Screen - Identifying new Hits for Kinase and GPCR Drug Targets
MolSoft's ICM accurate flexible docking, cheminformatics and virtual screening technology provided the unique and powerful engine for a successful screen of as many as 11 billion chemical compounds, potential drug candidates. The USC laboratory of Prof. Vsevolod Katritch report in Nature an approach called V-SYNTHES. The tool performs a ICM-docking based hierarchical structure-based screening of a Enamine REAL Space giga library. The screen resulted in the discovery of new lead compounds for the GPCR CB2 and the Kinase ROCK1.
For many years, MolSoft has been developing methods for fast accurate docking and scoring tools, and incorporating those tools to search giga-sized libraries. These tools include, ICM-Pro docking, GIGA search, and a shape property-based pharmacophoric search method called RIDE. GIGA search and RIDE were applied to the NIH SAVI database and Enamine REAL database.
For V-SYNTHES, ICM-Pro was used as the docking engine as well as for the preparation of the reaction libraries and enumeration of the giga-sized virtual databases. Docking and screening simulations were then undertaken using MolSoft's ICM-Pro Virtual Ligand Screening tools. The docking uses MolSoft's Biased Probability Monte Carlo method, optimized force field, docking score to dock and score fragments of a combinatorial library, and then an empirical score is derived and applied to a much larger library. At the end, the top scoring hits from the library are re-docked and scored again. This hierarchical protocol accelerates the docking and scoring run and makes it applicable to very large databases. The flexibility of the binding pocket was represented using MolSoft's 4D docking methodology which uses an ensemble of structures efficiently for fast and accurate flexible-receptor ligand docking.
Prof. Katritch at USC says "We are very excited to use the MolSoft ICM-Pro technology to address the challenges of screening very large chemical databases for the discovery of drug candidates."
You can view and download additional ICM scripts used for this work at the Katritch lab GitHub site which are ready for use with the ICM-Pro + VLS desktop modeling software.
Please contact MolSoft (www.molsoft.com info@molsoft.com) if you have any questions about how to apply this approach to your drug discovery program.
Sep 24th 2021: MolSoft's AI and Structure Based Drug Design technology supports two new Oncology Focused Companies Gwynant Therapeutics and Celyn Therapeutics
May 6th 2021: MolSoft-NIH Collaboration makes Billion Chemical SAVI Database Available for Screening in ICM
MolSoft has collaborated with the NIH/NCI (Tarasova and Nicklaus labs at NIH/NCI) to make their Synthetically Accessible Virtual Inventory (SAVI) database available to screen using MolSoft's Rapid Isostere Discovery Engine ( RIDE). You can read more about how the NIH developed SAVI in this publication here. The SAVI database contains more than 1 billion chemicals and RIDE can screen 500K chemical conformations/sec/GPU so the database can be screened very quickly.
RIDE is a fast 3D molecular similarity search method based on Atomic Property Fields (APF). RIDE searches databases of compound conformers for molecules that are isosteric to the query, i.e. have similar 3D configurations and distributions of atomic properties. RIDE is a multi-conformational optimal 3D superposition in which both the query molecule and the molecule in the database are represented in multiple low energy conformations to reflect the flexible nature of both molecules. RIDE is available in the ICM-Pro + VLS package.
You can download the SAVI database and other databases formatted for RIDE here http://www.molsoft.com/addons/conf/ Please be aware that the SAVI database is 4.3 Terabytes in size so will take a while to download and we recommend for optimal performance the database should be located on local SSD or fast HDD.
April 29th 2021: MolSoft provides structure-based drug design support to three new oncology focused companies Lomond Therapeutics, Eil Therapeutics and Ness Therapeutics and an inflammation focused company Vyrnwy Therapeutics
January 15th 2021: MolSoft and Mcule Collaborate to Connect the ULTIMATE database with MolSoft's ICM Drug Discovery Suite
Mcule's ULTIMATE library is now available in Molsoft's ICM-Chemist and ICM-Pro desktop modeling packages. ULTIMATE contains more than 100 million drug-like chemicals which can now be searched using 2D and 3D pharmacophore methods developed by MolSoft. As more than 95% of the compounds in ULTIMATE are completely novel, researchers can easily find new chemical starting points and move away from patent space.
ICM allows users to search ULTIMATE using MolSoft's MolCart Giga-Search technology. It can be searched using chemical substructure, patterns/fingerprints, similarity, exact match, and the results can be analyzed and filtered using Multi Parameter Optimization (MPO). Structure-based and fast ligand-based virtual screening of ULTIMATE can also be undertaken in the ICM-Pro software to discover new hits and scaffold hopping.
"Molsoft offers excellent tools for computational chemistry. We hope that this synergy will shorten the time of various Hit to Lead projects across the industry." - Robert Kiss, CEO of Mcule
"We are excited to collaborate with Mcule to make 100M potential drug candidates available in the ICM drug discovery suite. It gives our customers another fast way to discover novel chemistry and quickly get on the path to patentable chemicals. The interface inside ICM makes it easy for our customers to search and acquire new lead chemicals via Mcule's compound sourcing services." Andrew Orry Ph.D. Molsoft LLC
About Mcule:
Mcule is a chemical marketplace for drug discovery with services based around small-molecule compound sourcing. While the traditional Mcule database aggregates supplier catalogs to cover the in stock chemical space, ULTIMATE contains 122 million novel, synthetically accessible compounds. Mcule recently launched its custom synthesis auction site SynthAgora to cover chemical space not present in any catalogs.
About Molsoft:
Molsoft is a leading provider of software tools, databases and consulting services in the area of molecular modeling, cheminformatics and rational drug design. MolSoft offers complete solutions customized for a biotechnology or pharmaceutical company in the areas of computational biology and chemistry. ICM Chemist is a standalone suite of cheminformatics programs for chemical drawing and editing, chemical database generation, chemical searching, clustering, and enumeration. ICM-Pro is MolSoft's main desktop modeling software which can incorporate the tools in ICM-Chemist as well as molecular modeling, virtual screening, and machine learning drug target and ADMET models (MolScreen).
2020
October 7th 2020: MolSoft ICM Java Script: Share Fully Interactive 3D Molecules and Chemical Spreadsheets via a Web Browser
MolSoft is excited to announce the availability of new ICM Java Script (IcmJS) functionality and a convenient add-on for Google Drive. IcmJS is a JavaScript version of MolSoft's ICM Desktop Modeling software and brings the highest quality 3D graphics to the web. IcmJS allows you to view and share with your colleagues fully interactive 3D molecular structures and chemical spreadsheets and plots directly in a web browser. It contains a full suite of molecular visualization tools click here for more information.
Download the Google Drive App to see some examples.
An easy to use Google Drive app has been developed which makes file sharing and visualization easier. You can download IcmJS for Google Drive here.
Here are some examples for you to try:
Click here to see some examples using the Google Drive app (download the IcmJS app first).
Some of the new IcmJS include:
Open fully interactive 3D molecular files directly in your web browser.
Display and share docking hitlist and chemical tables click to view the docked ligand in the receptor, toggle on/off the display of multiple ligands.
Display chemical spreadsheets: sort columns, toggle 3D chemical display, view interactive plots, radar plots, pie charts
interact with chemical cluster trees
fully interactive dendrograms
Display interactive 2D ligand-receptor interaction diagrams linked to 3D structure
Display interactive alignments linked to 3D structure.
How to make and share the IcmJS files:
Open ICM software, prepare 3D molecules (make slides), tables or docking hitlist. For molecular graphics you can prepare slides in the free ICM-Browser.
File/Save as .icb format. This is Molsoft's ICM binary format.
Save the file to your google Drive.
How to view the 3D file:
Go to Google MarketPlace and download IcmJS (only has to be done once).
Click on the saved .icb file in Google Drive choose IcmJS as the app to view it.
Sept 1st 2020: Arrakis Therapeutics incorporate MolSoft's ICM PocketFinder and Docking Methods into their Small Molecule-RNA Disovery Platform
Arrakis Therapeutics report a new photoaffinity platform for the analysis of chemical interactions with RNA. The paper in ACS Chemical Biology shows how the method they developed called Photoaffinity Evaluation of RNA Ligation-Sequencing (PEARL-seq) can be used for RNA drug discovery by rapidly identifying ligand binding across many types of RNA. MolSoft's ICM PocketFinder method was used to generate an understanding of where small molecules might bind to Aptamer 21 and then ICM docking was used to predict the docked pose of a probe. The docked pose of the ligand was then used to generate hypotheses around which the binding affinity of the ligand could be improved. The SAR data shows that this method has utility for the design and optimization of new RNA-based small molecule therapeutics.
June 15th 2020: Press Release - Molsoft and Advent Informatics enter into a distribution agreement
San Diego, USA and Pune, India: Molsoft LLC, a leading software development company, announced today that they have entered into an agreement with Advent Informatics to promote distribution of Molsofts ICM computational drug discovery software in India.
Through this agreement chemists and biologists in India will be able to facilitate access to the software tools and services for lead discovery, ligand docking, screening, modelling, cheminformatics, bioinformatics offered from Molsoft LLC. Under the terms of this agreement Advent Informatics will utilize its marketing and research expertise in India to distribute and support Molsoft products in India.
"High quality modelling tools are very essential in drug discovery, we want to empower the research community in India by providing access to software that is used by leading global pharmaceutical companies" said Surojit Sadhu, Founder, Advent Informatics.
"We are excited to enter into this agreement with Advent Informatics because they have a lot of experience in the Indian IT drug discovery market. The ICM-Pro desktop modeling suite is currently outperforming other software for ligand docking and screening accuracy and working with Advent will help expand access to the software for scientists in India said Dr. Andrew Orry (MolSoft LLC).
About Molsoft LLC: Molsoft is a San Diego company that develops new breakthrough technologies in computational chemistry and biology. Molsoft is committed to solving intellectually challenging problems in drug discovery and computational biology. Molsoft has built proprietary computational environments including the ICM suite of programs for molecular modeling, bioinformatics, cheminformatics and ligand docking, and MolCart. Molsoft offers software tools and services in lead discovery, modeling, cheminformatics, bioinformatics, and corporate data management, forms partnerships with biotechnology and pharmaceutical companies, and is the recipient of several NIH and DOE-sponsored awards.
About Advent Informatics: Advent Informatics is a Pune based start-up focusing on providing state-of-the-art software to researchers and students. Advent Informatics promotes solutions from International companies by acting as a bridge between researchers based in India and international software and database companies. Primarily focusing on life sciences, we offer software and IT solutions that catalyse the research of users working in the field of Chemistry, Biology and Drug Discovery, as well as provide technical support offered to its users.
RIDE is a fast 3D molecular similarity search method based on Atomic Property Fields, developed at MolSoft. RIDE searches databases of compound conformers for molecules that are isosteric to the query, i.e. have similar 3D configurations and distributions of atomic properties.
GPU-based implementation is capable of searching ~0.5mln conformers/second on a single GPU card and perform 3D virtual screens of millions of compounds with the level of interactivity comparable to 2D searches. VLS benchmarking on DUDe indicates that RIDE searches produce enrichments on par with much slower standard flexible APF VLS.
Scaffold hopping - discover structurally novel chemicals based on a lead.
Hit follow-up - quickly discover new chemicals with similar 3D pharmacophoric binding properties to your lead.
A simple web-based interface is provided for the ease of use by chemists, while batch script may be used by chemoinformatitians. In either format, RIDE allows fine-tuning of the queries to generate most desirable hit lists. This includes:
Atom Weighting - Contributions of different portions of the molecule can be modulated with per-atom weights to reflect relative importance of certain moieties.
Excluded Volumes meet Shape Matching - An envelope penalty can be applied to the regions that surround all or part of the query molecule to prioritize hits without bulky extensions in constrained regions.
January 1st 2020: Macrocycle Modeling - MolSoft ICM-Dock Again Performs Well in Industry-Wide Grand Challenge Competition.
The results of the "blinded" D3R Grand Challenge 4 ligand docking competition have been published and MolSoft once again has outperformed a range of other methods for ligand binding pose and activity prediction.
The work led by Max Totrov Ph.D. (Principal Scientist, MolSoft) has been preented at the D3R evaluation meeting and published in the Journal of Computer Aided Molecular Design (see publication here ). The results from the D3R GC4 are presented along with an analysis of ICM performance in a large (246 complex) macrocycle docking benchmark. Sampling of flexible rings is directly incorporated into the ICM docking and the paper describes the best practices for macrocycle docking.
ICM accurately predicted the pose of BACE macrocycle inhibitors to within sub-angstrom accuracy using multiple receptor and ligand-biased docking. ICM was ranked top in the cognate receptor part of the competition where the organizers provided the protein structure bound to protein. Notably, unlike other top ranking methods, for our submission in this subcategory we have not used any ligand template information.
MolSoft's 4D multiple receptor conformation docking method for incorporating induced fit in the binding pocket was used to dock the macrocycles. The method was improved from GC2 (Lam et al 2018) and GC3 (Lam et al 2018 ) by introducing scoring/energy offsets that optimize the choice of receptor conformation used. In the cross-docking stage of the challenge and when applicable the docking was guided by 3D pharmacophore using MolSoft's Atomic Property Field (APF) method (Totrov 2008).
MolSoft would like to thank the organizers for coordinating the D3R challenge and providing a valuable way of independently assessing the performance of the ICM-Docking and 3D QSAR tools.
2019
August 26th 2019: American Chemical Society Meeting 2019
MolSoft's founder Prof. Ruben Abagyan will be presenting at the 2019 American Chemical Society Annual Meeting in San Diego. The title of his talk is "Multi-target pharmacology of kinase inhibitors, beneficial off-targets and allosteric sites". If you are at the meeting the talk will on Tuesday August 27th at 10:15 AM at the OMNI San Diego hotel.
June 20th 2019: MolCart Giga Search - World's first Billion Chemical Substructure Search Method
MolSoft is excited to announce the release of MolCart Giga Search - the first of its kind substructure method for efficiently searching databases of 10^9 chemicals in size. Read more about the method here on MolSoft's website here.
Databases of synthesizable and virtual chemicals are getting larger each year, therefore the ability to efficiently mine them is critical for drug discovery projects. MolCart Giga Search helps drive your SAR search by providing a way to build target-specific libraries, find derivatives and homologs and screen using SMILES and SMARTs and other chemical properties.
The latest release of MolSoft's ICM-Chemist software provides an easy to use Graphical User Interface for substructure searching of large chemical databases. You can search billions of chemicals or your own custom database using a combination of MolCart Giga Search and ICM-Chemist. Searches can also be scripted and there are large number of post-search processing tools such as Multi Parameter Optimization built into the software.
MolSoft recently collaborated with Enamine to implement MolCart Giga Search on the REAL database (720 million chemicals) you can use it here
Please contact MolSoft for more information about MolCart Giga Search - info@molsoft.com 858-625-2000.
March 7th 2019: Protein Structure and Function Exploration (PSAFE) - Using MolSoft's ICM for Teaching Undergraduates
Using ICM for Education - A collaboration between MolSoft and Prof. Charles Grisham at Univeristy of Virginia see publication here: https://t.co/WmdbL97HM4 see the PSAFE resource here: https://t.co/Se968FRgFU
February 7th 2019: Webinar Series Part 1 - Molecular Graphics, Protein Structure and Sequence Analysis
Thank you very much for your support of MolSoft's webinars. The first webinar series of 2019 attracted more than 650 participants over the course of the five weeks. You can watch the webinars in our Webinar Archives on YouTube or by clicking the links below.
How to Make Publication Quality 3D Molecular Images and Videos Webinar Recording
How to Embed Fully Interactive 3D Molecules into a Web Page or MS PowerPoint Webinar Recording
Protein Structure and Crystallographic Analysis Tools Webinar Recording
ICM-Bio: Linking Protein Structure, Sequences and Alignments. Webinar Recording
How to Identify Ligand Binding Pockets and Allosteric Sites in Protein 3D Molecules. Webinar Recording
2018
September 11th 2018: MolSoft publishes Protein-RNA docking method
Click here to read paper by Arnautova et al in J Chem Theory Comput. 2018. Paper Abstract: Protein-RNA interactions play an important role in many biological processes. Computational methods such as docking have been developed to complement existing biophysical and structural biology techniques. Computational prediction of protein-RNA complex structures includes two steps: generating candidate structures from the individual protein and RNA parts and scoring the generated poses to pick out the correct one. In this work, we considered three recently developed data sets of protein-RNA complexes to evaluate and improve the performance of the FFT-based rigid-body docking algorithm implemented in the ICM package. An electrostatic term describing interactions between negatively charged phosphate groups and positively charged protein residues was added to the energy function used during the docking step to take into account the greater role that electrostatic interactions play in protein-RNA complexes. Next, the docking results were used to optimize a scoring function including van der Waals, electrostatic, and solvation terms. This optimization yielded a much smaller weight for the solvation term indicating that solvation energy may be less important for the scoring of protein-RNA structures. Rescoring of the generated poses with the new scoring function led to much higher success rates, while pose clustering by contact fingerprints produced further improvements, achieving a success rate of 0.66 for the top 100 structures.
September 7th 2018: MolSoft Outperforms a Range of other Methods for Docking Pose and Affinity Prediction Accuracy in D3R Grand Challenge 3
Scientists at MolSoft led by Max Totrov Ph.D. participated in the Drug Design Data Resource (D3R) Grand Challenge 3 (GC3) and once again achieved great success in the accuracy of docking pose prediction and ligand binding affinity. The competition is a "blinded" open challenge for the worldwide computational chemistry community bringing together teams using different docking methods and software.
The results and methodology used by MolSoft in GC3 are published in a recent article by Lam et al entitled "Hybrid receptor structure/ligand-based docking and activity prediction in ICM: development and evaluation in D3R Grand Challenge 3". Also, you can watch a video recording of Max Totrov's presentation on the results at the D3R workshop here.
MolSoft's submissions ranked first place for docking pose and affinity prediction for all targets according to key criteria and builds upon MolSoft's successful performance in Grand Challenge 2 (GC2) - see Lam et al J Comput Aided Mol Des. 2018 32:187 "Ligand-biased ensemble receptor docking (LigBEnD): a hybrid ligand/receptor structure-based approach"). In GC2 Molsoft ranked first place for docking accuracy and binding energy prediction.
Using MolSoft's ICM-Pro software the team participated in pose and ligand affinity prediction challenges for six different targets including Cathepsin S and five different kinases. The predictions were made using a combination of ICM-docking biased by the Atomic Property Fields (APF) of known ligands acting as templates to guide the pose prediction. The image on the right shows the APF fields for ligand bias for Cathepsin S. This approach provides a way of combining a physics-based docking score as well as a phenomenological APF pseudo energy score for pose ranking. For pose prediction ICM returned a median RMSD for the top pose of 1.7≈ and a median RMSD of 1.06≈ for best pose of five (Figure 1). The disparity between the average RMSD for top pose and the average RMSD for the best pose of 5 is most likely due to crystal contact effects which were not taken into account in the analysis of the results.
Figure 1: MolSoft's performance in the Pose Prediction challenge of GC3 is highlighted by a red arrow.
For the affinity prediction targets it was important to incorporate flexibility of the binding pocket. This was achieved by using pre-aligned PDB structures for each target from the Pocketome database and then selecting up to 10 representative X-ray structures along with compatible ligands used as APF templates.
In the affinity prediction challenge the docked poses of the ligands were scored using a hybrid approach, machine learning (PLS and RFR) on a combination of Receptor/Physics based terms and Ligand/APF-based 3D chemical similarity terms (APF/P 3D QSAR). Evaluated by Kendall tau ranking correlation across all ligands, ICM ranked first place in affinity prediction for all targets (Figure 2).
Figure 2: MolSoft's performance for four targets for the prediction of ligand affinity. Red arrow points at Molsoft's submission.
June 28th 2018: MolSoft's ICM Ranks First in Independent Covalent Docking Tools Comparison
A recent publication by Scarpino et al compared the performance of six covalent docking methods. They selected a large and divergent set of covalent complexes and assessed how well each software could achieve the crystal structure pose of the covalent ligand.
At a two Angstrom RMSD cut off MolSoft's ICM-Pro software was able to achieve the experimental ligand pose in the Top 1 conformations in 62% of the cases and in the Top 10 conformtions in 88% of the cases. MolSoft's ICM-Pro software outperformed the other programs in all tests including reproducing the experimental binding modes and scoring. ICM also performed well with more challenging flexible ligands with 35 heavy atoms or more. In an interesting side test described in the paper, it was found that ICM-Pro's non-covalent conventional docking procedure also gives a remarkable rate of accurate predictions for covalent ligands to modified receptors (CYS/ALA mutations- 86% near native).
The figure below (from Scarpino et al) shows the re-docking success rate at top 1 pose (dark colors) and top 10 poses (lighter colors).
April 3rd 2018: MolSoft to Present Short Course at Drug Discovery Chemistry Conference in San Diego
Max Totrov (MolSoft LLC) will present at the "Trends in Physical Properties of Drugs" short course at the Drug Discovery Chemistry Conference in San Diego on Apil 4th. The course topics include:
Properties important for enhanced efficacy, delivery, and formulation
pKa, tautomerism, crystallization, others
Computational prediction: What works - what does not
February 22nd 2018: Max Totrov Ph.D. (Principal Scientist, MolSoft) to present at D3R workshop.
Today, Maxm Totrov Ph.D. will present the results of MolSoft's excellent performance in the docking D3R docking challenge at the D3R Workshop. MolSoft's ICM software ranked in first place for average RMSD docking accuracy in the 2016-17 Drug Design challenge competition http://drugdesigndata.org/ (D3R). This work was recently published in the Journal of Computer Aided Molecular Design (see Lam, Abagyan and Totrov 2018).
Totrov: Abagyan group has a useful tool called Pocketome that can retrieve and score X-ray structures consistent with ligands. #D3R2018pic.twitter.com/JfGiSUu3hS
September 7th 2017: FDA approves Vabomere: structure-based design using Molsoft's ICM was key to its discovery.
On August 29th 2017 the U.S. Food and Drug Administration (FDA) granted approval of Vabomere to RemPex Pharmaceuticals (part of the Medicines Company ). Vabomere is a new antibacterial drug for the treatment of complicated urinary tract infections which is a life threatening condition (click here for FDA Press Release).
Vabomere is a combination of meropenem (previously developed carbapenem antibiotic) and vaborbactam (RPX-7009), a novel cyclic boronate beta-lactamase inhibitor. Structure-based design by MolSoft's scientists (lead by Maxim Totrov Ph.D.) using the ICM-Pro desktop modeling software was key to the discovery of RPX-7009. The novel cyclic boronate chemotype was first modeled in-silico at Molsoft and prioritized for synthesis based on modeling and docking into several beta-lactamase target enzymes.
The work leading up to this discovery was published by RemPex and MolSoft in the Journal of Medicinal Chemistry - you can read more here.
MolSoft ICM D3R Docking Challenge Success
MolSoft's ICM software ranked in first place for average RMSD docking accuracy in the 2016-17 Drug Design challenge competition http://drugdesigndata.org/ (D3R).
Along with over 50 other participants, the Molsoft group led by Maxim Totrov Ph.D.(Principal Scientist, MolSoft), submitted blind docking pose predictions for the Farnesoid X receptor (FXR) which is a drug target for dyslipidemia and diabetes. The MolSoft team used the pocketome (www.pocketome.org) entry for FXR and the ICM-VLS, Atomic Property Fields (APF) and machine learning methods in the ICM-Pro software to predict the interaction of 36 FXR ligands and the binding affinity of 102 other ligands.
In January 2017 the organizers (D3R) distributed the evaluation results which showed that MolSoft's submission ranked first in RMSD for the top scoring pose and was the only one with average RMSD below 2.0A (Figure 1 below). In the Binding Energy Prediction competition ICM APF dock and icm-MMGBSA method ranked in first place and produced the lowest RMSE for Stage 2 Set 1 set of Kd values for 15 ligands (Lam et al 2018).
August 28th 2017: Free Webinar Series Presented by MolSoft
January 31st 2017: Virginia Tech and MolSoft awarded NIH grant to Refine Malaria Drug
2016
November 10th 2016: MolSoft Release ICM Version 3.8-5.
MolSoft is excited to release a new ICM version 3.8-5. Please click here to read more about the new features.
June 14th 2016: New Ligand Interaction Diagrams
Now you can generate a 2D interaction plot of a ligand with the binding pocket (see image on the right). The image is annotated with hydrogen bonds interacting residues. The residue interactions surface and proximity are represented by the size of the residue label and distance respectively. Grey parabolas and broken thick lines indicate solvent accessible regions and the ligand is shaded by property. [Documentation]
A guide to the coloring and representation of the 2D diagrams:
Green shading represents hydrophobic region.
Blue shading represents hydrogen bond acceptor.
White dashed arrows represents hydrogen bonds.
Grey parabolas represents accessible surface for large areas.
Broken thick line around ligand shape indicates accessible surface.
Size of residue label represents the strength of the contact.
2D distance between reidue label and ligand represents proximity.
June 7th 2016: MolSoft ICM now supports MMTF Format.
The Macromolecular Transmission Format (MMTF) is a new compact binary format to transmit and store biomolecular structural data quickly and accurately. MMTF is now supported in the latest ICM version it does not have any limitation on number of atoms and is easier to use than the original PDB. You can read more about MMTF here at the PDB website.
The image on the left shows the entire HIV-1 capsid (~2.4 million atoms PDB:3j3q).
In ICM (v3.8-5 and higher) use the command below to read in and display a mmtf file.
readpdbmmtf
Apr 22nd 2016: MolSoft Releases Fast High Quality JavaScript 3D Molecule Viewer
IcmJS (formerly known as ActiveIcmJS) is a JavaScript/HTML5 viewer for 3D Molecular Graphics which does not require any plugin or installation. It runs on all modern browsers including Chrome, Firefox and Safari and is also mobile device friendly. IcmJS gives you full access to the ICM shell and graphics on a web browser. This means that commands available in the free ICM-Browser are also available on the web via IcmJS.
You can view some IcmJS examples here:
WONKA: objective novel complex analysis for ensembles of protein-ligand structures (SGC Oxford ).
OOMMPPAA: a tool to aid directed synthesis by the combined analysis of activity and structural data (SGC Oxford ).
ActiveICM Reference: Raush, E., Totrov, M., Marsden, B. D. & Abagyan, R. A new method for publishing three-dimensional content. PloS One 4, e7394 (2009).
Want to Learn More? Read more on our IcmJS webpage or register for the IcmJS Webinar here.
Mar 13th 2016: Ruben Abagyan (MolSoft Founder) will present at the ACS Meeting in San Diego this Weekend.
Prof. Ruben Abagyan (MolSoft Founder and UCSD) will present at talk at the ACS meeting entitled "Toward predictive structural polypharmacology via flexible docking of ligands to the organismal pocketomes".
Abstract :Identification of molecular targets and pathways affected by drugs, environemtal compounds or metabolites is one of great challenges of computational structural biology. A number of methods addressing the problem at the level of chemistry-based machine learning have been developed. We attempted to develop an approach that would be based on a complete collection of crystallographically determined pockets, the Pocketome, complemented with the co-crystallized ligands. The newest release of the Pocketome contains around two thousands ensembles that can be converted to predictive models, and futher characterized by the noise distribution needed to derive the probability values, as well as, in some cases, additional training sets with pKd, IC50 or other activity data. Each new molecule is docked into those pockets or cumulative pharmacophoric fields and scored according to the model. We demonstrate how this system identifies previously unknown targets associated with adverse effects of some drugs and environmental compounds, as well as repurposing drugs to a different indication. A larger evaluation of this platform is underway a group of computational researchers from Novartis.
Feb 3rd 2016: MolSoft Publishes the Agenda for the 2016 ICM User Group Meeting
Please join us for MolSoft's ICM User Group Meeting 2016 to be held in San Diego on March 17-18. Everyone is welcome to attend and we have exciting speakers from academia and the biotech/pharma industry. You can view the meeting schedule here.
Date: March 17-18 2016
Location: MolSoft 11199 Sorrento Valley Road, S209 San Diego CA 92121 [ map ] [ hotels ]
Registration: $99 click here to download a registration form or call 858-625-2000 x108.
Schedule: Click here for full meeting agenda (subject to change)
UGM Archives: See photos and content from previous User Group Meetings here.
Speaker topics will include:
Latest Developments at MolSoft and new features in the pipeline
MolScreen: A set of high quality models for a broad range of pharmacology and toxicology targets
A new paper in Nature Chemical Biology describes the discovery of a potent small molecule which has the potential to be used to treat Spinal Muscular Atrophy (SMA) a debilitating motor neuron disease. The disease is caused by a deficiency of Survival of Motor Neuron (SMN) gene which is the most common cause of mortality in children. The pyridazine class of compounds described in this paper enhance SMN2 splicing which elevates full-length SMN protein. It is proposed that the small molecule modulators enhance binding of U1 snRNP to the SMN2 exon 7 5'ss.
Using MolSoft's ICM Alibero method , scientists at Novartis Institute of Biomedical Research were able to model the binding mode of the molecule with RNA (See Figure- left). To model the flexibility of RNA a population of 150 RNA models was generated and the Alibero method was able to determine the model that best separated known binders from non-binders. On the basis of this model they were able to hypothesize that the small molecules enhance the stabilization of the RNA duplex in the 1A bulge major groove.
June 11th 2015: MolSoft and Novartis Collaborate to Develop FOCUS - A global communication and modeling platform for medicinal chemists.
MolSoft and Novartis have developed a new desktop modeling and communication environment for drug discovery called FOCUS based on MolSoft's Internal Coordinate Mechanics (ICM) software. The FOCUS platform is described in a publication in the ACS Journal of Chemical Information and Modeling (see Stiefl et al 2015). FOCUS is a platform that helps users communicate chemical and structural data, develop new ideas and support decision making during the drug design cycle. FOCUS can be integrated into an informatics and high performance computing environment giving the user a single interface to many capabilities.
FOCUS was recently shown being used by Novartis scientists in a PBS documentary called "Cancer: Emperor of all Maladies". The image shows interaction of Novartis' cancer drug LDK378 with ALK.
FOCUS is the result of more than eight years of collaboration between MolSoft and Novartis. Globally at Novartis there are around 1500 registered users and it has become a popular tool for medicinal and computational chemists as well as structural biologists and bioinformaticians. The core functions of FOCUS can be found in MolSoft's ICM-Chemist-Pro desktop modeling and cheminformatics software product (http://www.molsoft.com/icm-chemist-pro.html). FOCUS contains a wide range of applications including a comprehensive set of feature-rich cheminformatics tools and a 3D ligand editing and design hub. Using MolSoft's ICM scripting language Novartis created customized interfaces that enabled them to build fully interactive html pages, menus, dialogs, panels and table actions all linked to internal and external service calls. The FOCUS development team paid particular attention to make it user-friendly for non-computational researchers.
The Novartis FOCUS development team worked closely with MolSoft's software engineers to build a chemically intuitive and user friendly ligand design interface called the ICM Ligand Editor. The Editor is integrated into FOCUS as well as MolSoft's ICM-Pro and ICM-Chemist-Pro desktop modeling and cheminformatics products. Using the editor, a ligand bound to a protein can be modified in 2D or 3D and the effects of the modification on binding can be immediately calculated and observed. Modifications are stored in the memory, which enables full undo and redo capabilities and the medicinal chemist can conveniently store new designs in fully interactive 3D spreadsheets. Rich graphics tools are available for the representation of the binding pocket surface and interactions between the receptor and ligand. The editor is powered by MolSoft's highly acclaimed ICM docking software and contains options for constrained, induced fit, fragment, and covalent docking. The ligand editor can also be used for 3D pharmacophore modeling using MolSoft's Atomic Property Fields.
Screenshot of the ICM Ligand Editor which is incorporated into FOCUS. The ligand is displayed in 3D on the right hand side and the "one-click" editing tools are on the left panel.
The publication describes the development steps that Novartis undertook in the development of FOCUS along with advice for people who want to build a similar platform for their company. If you are interested in finding out more please contact MolSoft (info@molsoft.com) or call 858-625-2000 x108.
April 2nd 2015: MolSoft ICM Software shown in PBS Documentary - Cancer: The Emperor of all Maladies
Last night's PBS documentary "Cancer: The Emperor of all Maladies" showed ICM in action (Jump to 15min 24secs). Scientists at Novartis are shown in the documentary using ICM and the 3D Ligand Editor to visualize the interaction of the drug LDK378 which is a selective inhibitor of ALK. ALK is a target found in a variety of cancers including lung cancer.
Click to Play Video:
2014
December 10th 2014: MolSoft will host the ChEMBL workshop on Jan 28th 2015
MolSoft is excited to announce that we will host the San Diego leg of the ChEMBL USA tour on January 28th 2015. Please RSVP here.
About the Workshop:
Every year, research produces a vast amount of data describing the biological effects of chemical substances. This valuable information, while public, is usually in a form not accessible for systematic data extraction (data mining) and lacks consistent standardization.
ChEMBL is an Open Data database that contains this information manually extracted from the primary scientific literature. The database contains binding, functional and ADMET information for a large number of drug-like bioactive compounds. Data is further curated and standardized (assay read outs and chemical structures) to maximize their quality and utility across a wide range of chemical biology and drug-discovery research problems.
We have recently complemented ChEMBL with SureChEMBL, a web-based daily updated database of full patent text and automatically extracted compound structures. This allows advanced chemistry based querying of the patent literature. We will present an overview of both resources, and then show specific use common usage and data analysis scenarios
December 3rd 2014: Ruben Abagyan (MolSoft Founder) gives Keynote Speech at the In Silico Drug Discovery Conference
Dr. Ruben Abagyan was the keynote speaker at the In Silico Drug Discovery Conference held in Durham NC from Dec 3-4. Dr. Abagyan's talk was entitled "Drug Discovery with Three Dimensional Models of Everything".
September 1st 2014: New ICM Version 3.8-1 Released
You can download the new version of ICM from our Support Site.
New features include:
Modeling
MERSi: Mutation Effects with Relaxation of Side-chains in Internal coordinates.
Build homology models with rigorous refinement in batch (see Homology/Batch Build and Refine).
Loop Grafting - transfer a loop region from one protein to another. Documentation
Build peptides and make mutations using unusual amino acids. A table of 250 non-standard Amino Acids added and enabled for the mutation tool.
Protein Sculpting can be used to model the interaction of two connected domains. You simply drag the molecules around a hinge region followed by a minimization. Documentation
Sequence and Alignment
Calculate Global or Local Sequence Similarity and Identity in an Alignment. Documentation
Sequence drag'n'drop between sessions is now supported.
Multi-line sequence alignment annotations are now supported.
Cheminformatics
Extensive chemical dictionary is now available in the molecule editor.
Direct search of the Crystallography Open Database (COD). The COD is an open-access collection of crystal structures of organic, inorganic, metal-organic compounds and minerals, excluding biopolymers.
Direct interface in GUI for searching PubChem. Documentation
Match chemical 2D sketch to PubChem and extract PubChem CID or ID number (Use Insert Column and choose Function Chemical PubChem).
New set of pre-prepared common covalent binding mechanism reactions for covalent ligand docking. Documentation
See the Release Notes for a full list of new features and bug fixes.
June 4th 2014: Introducing MERSi: Mutation Effects with Relaxation of Side-chains in Internal Coordinates
MERSi is a new algorithm available in ICM-Pro that allows you to predict the effect of a mutation upon protein stability and protein-protein binding free energy.
Predict Change in Binding Free Energy upon Mutation
The binding free energy change ΔΔGbind, is computed as a difference between the binding free energy of mutant and wild type complexes. To account for structure relaxation upon mutation, side chain re-packing in the vicinity of the mutated residue by a Monte Carlo simulations carried out. "Scan Protein Interface" option allows to mutate all residues (one by one) of the Interacting Part in close contact with the second part of the complex. Documentation | Tutorial
Predicting Change in Protein Stability upon Mutation
The stability change is calculated as a weighted sum of a complete set of physically meaningful free-energy contributions, including van der Waals attractions, repulsions, electrostatics, hydrogen bonding, and solvation term. Residue-specific constants that account for the free energies of the unfolded and misfolded states were derived empirically using a large set of experimental data. Mutation of a given residue is followed by Monte Carlo simulations with flexible side chains for the mutated residue and its neighboring residues. Backbone flexibility is also considered for special cases such as proline and disulfide-forming/breaking cystein mutations. Documentation
February 14th 2014: UK Scientists rank iMolview the best iPad/Android Molecular Viewer
iMolview came out on top in a recent comprehensive review of 12 molecule viewing apps for iPad and Android devices. The review article called "High-Quality Macromolecular Graphics on Mobile Devices: A Quick Starter.s Guide" was published in the Methods and Molecular Biology series by Drs. Yiu and Chen from King's College London, UK. Please click here to download the paper (subscription maybe required).
The article states that iMolview "tops the list because of its user friendliness and it offers the most complete set of functions for visual communication". The paper also includes a useful iMolview tutorial which takes you through the steps of importing molecules, coloring, transparency, surfaces, and exporting images.
February 5th 2014: MolSoft Releases New ICM Version 3.8
MolSoft is excited to release a new version of ICM. Please download the new version from www.molsoft.com/download.html. The Release Notes contain a complete list of new features and fixes but the key ones are listed below.
Graphics
Anaglyph 3D can be viewed using low cost 3D glasses and without any expensive 3D compatible hardware or monitors. It can be viewed directly on your computer's monitor, conference room projector, or on your iPad or android device. You can also print the image out rendered for viewing in 3D. Any red and cyan glasses will work well. [More Information] and [Documentation]
MolSkin MolSkin is a new macro that instantly makes publication quality graphics for surfaces and colors contact patches. [Documentation]
Movies from Slides A simple way to make a movie is by making a set of slides containing the scenes and then let ICM package them into a movie file. [Documentation]
Docking and Screening
SCARE The SCARE method allows you to take into account induced fit of the receptor upon ligand docking. The method systematically scans pairs of neighboring side-chains and replaces them with Alanine and docks a ligand to each of the "gapped" models. A full description of this method and validation is published here. [Documentation]
MolScreen is a set of high quality 2D fingerprint and 3D pharmacophore models for a broad range of pharmacology and toxicology targets.
The models can be used for lead discovery or counter screening. The models use MolSoft's 2D QSAR/Fingerprint and 3D Atomic Property Fields ( Totrov 2008) methods. [More Information] and [Documentation]
Fragment Screening Screen a database of chemical fragments which are then ranked by p-value providing a quantitative measures of confidence that a fragment is a true active and binds in the specific pose in absolute terms, instead of just ranking it versus other fragments. [Documentation]
Docking directly from table The ability to dock directly from a loaded chemical table gives you some flexibility about which chemicals you wish to dock from a table and makes docking more interactive and more convenient. [Documentation]
New Data Search and Retrieval Options
Blast Search Now you can BLAST search the NCBI database directly in the ICM GUI. [Documentation]
Pocketome Search The Pocketome (www.pocketome.org) is an encyclopedia of conformational ensembles of all druggable binding sites that can be identified experimentally from co-crystal structures in the Protein Data Bank. [Documentation]
Drugbank Search The DrugBank (http://www.drugbank.ca/)database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. [Documentation]
UniProt Search The Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data. [Documentation]
Cheminformatics
Fingerprints Extended Connectivity Fingerprints (ECFP) have been added.
Chemical Search Chemical Search dialog allows to select an existing chemical table for Molcart or local search (see image).
InChI is now supported.
ToxScore A ToxScore function is now available in the Insert Column Dialog. [Documentation]
Atomic Property Fields A chemical table can now be clustered by Atomic Property Fields (see Chemistry/APF Tools}
Export to Excel The 2D chemical sketch is now exported to Excel. [Documentation]
January 24th 2014: Introducing MolScreen - A Panel of >360 High Quality Screening Models
MolScreen is a set of high quality 2D fingerprint and 3D pharmacophore models for a broad range of pharmacology and toxicology targets. The models can be used for lead discovery or counter screening. The models use MolSoft's 2D QSAR/Fingerprint and 3D Atomic Property Fields ( Totrov 2008 ) methods.
The models can be screened directly using MolSoft's ICM-Pro + VLS software. Alternatively we can screen a set of chemicals for you via our contract research services. Please contact us for more information about how to use MolScreen.
December 19th 2013: Anaglyph 3D now available in ICM and iMolview.
MolSoft is excited to announce that Anaglyph 3D is now available to use in ICM and iMolview. Anaglyph 3D can be viewed using low cost 3D glasses and without any expensive 3D compatible hardware or monitors. It can be viewed directly on your computer's monitor, conference room projector, or on your iPad or android device. You can also print the image out rendered for viewing in 3D.
Here are the glasses the developers and scientists at MolSoft are using but any Red and Cyan 3D glasses will work.
To use Anaglyph 3D in ICM you will need the very latest version of ICM 3.7-3c and the documentation is here. Please contact us if you do not have the latest version yet. For mobile and tablet users please download the next iMolview update.
December 11th 2013: MolSoft ICM used in World's Largest Ever Virtual Screen Resulting in 3 New Lead Compounds.
At the recent Amazon Cloud Computing conference ( AWS Re:Invent ) scientists at Novartis reported using MolSoft's ICM-VLS in the largest ever virtual screen. MolSoft's software developers built a cloud-version of ICM-VLS which enabled Novartis to undertake this ground breaking achievement in cloud-based virtual screening. Cyclecomputing (a cloud vendor service company) enabled the jobs to be distributed to more than 30,000 cores in the USA and Europe. Novartis undertook 10.6 years of drug compound computations in 9 hours! The virtual screen resulted in three new lead compounds which are now being optimized by Novartis. This work was previously reported at MolSoft's user group meeting earlier this year and at BioIT 2012.
Watch a Video - click on image on right.
MolSoft's ICM-VLS has a strong track record of achieving high quality leads in ligand- and structure-based drug discovery. Please contact MolSoft if you would like to learn how you can undertake a similar screen.
August 29th 2013: MolSoft wins Competitive Bid to Work with AMG and TB Alliance on the Discovery of new TB Drugs.
MolSoft is excited to announce that they have won a competitive bid to work with AMG and TB Alliance on the development of vital new medicines that can be used to treat Tuberculosis (TB).
TB is a common disease which typically affects the lungs and if not treated correctly can be fatal. The current TB treatment is very long and can result in the patient not adhering to the strict regimen. With the recent approval of Bedaquiline as a treatment for multi-drug-resistant TB it gives hope that new and safe TB drugs can be identified.
In this collaboration, MolSoft will develop ligand-based models using published TB drug activity data as well as internal TB Alliance high throughput screening results. All work will be performed using MolSoft.s Internal Coordinate Mechanics (ICM) molecular modeling and cheminformatics software package which is used worldwide by academic and commercial laboratories. MolSoft will develop ICM 2D fingerprint and 3D Atomic Property Field (APF) models for each of the activity series. The 2D approach uses MolSoft.s proprietary method for generating hashed chemical fingerprints used in chemical search and similarity calculations. The 3D APF method, developed by Molsoft.s Principal Scientist Dr. Maxim Totrov, is a 3D pharmacophoric potential implemented on a continuously distributed grid which can be used for ligand docking and scoring.
Dr. Ruben Abagyan (MolSoft.s Founder and Professor at Skaggs School of Pharmacy and Pharmaceutical Sciences UCSD) says "The collaboration between chemists at TB Alliance and the scientific team at MolSoft represents an exciting application of their ICM ligand-based modeling and screening methods. MolSoft.s scaffold hopping technology is being used successfully in the pharmaceutical industry and it is a sign of this achievement that MolSoft was chosen to work on this project. I am confident that this alliance will give the best chance of identifying a new class of TB drugs."
Once the models have been prepared ICM-VLS will screen >12 million drug-like compounds to each model. High quality novel hits will then be chosen which are suitable according to the project.s pharmacokinetic and cytotoxicity criteria for hit to lead progression.
May 28th 2013: MolSoft Releases an HTML5 Molecular Editor
MolSoft announces the release of a HTML5 Molecular Editor which allows you to draw 2D chemical structure inside the web browser's page. It does not require
any plug-ins (like Java) and works in any modern HTML5 compatible browser with Java Script enabled (including mobile platforms). You can sketch chemicals as well as import and export in MOL or SMILES format.
The editor is made available free of charge to academic institutions, please
contact us if you wish to use it for commercial use. Please click here for more information on how to use the editor in your application.
To see the HTML5 Molecular Editor in action please visit one of our free popular online services.
May 27th 2013: New iMolview iPhone/iPad (v1.7) and Android (v1.2) Released
The biggest new addition to this version is the ability to read and contour electron density maps.
iMolview v1.7 iPhone/iPad
Tools/Load Electron Density Map
Loads electron density map from Uppsala EDS server (http://eds.mbc.uu.se/eds/). Once map is loaded you can toggle its display using 'Misc' panel in the main menu. In addition map can be contoured around selected atoms:
Select part of the molecule
Click on the contour icon in the 'Misc' panel on the pain menu.
Contouring level can be adjust using slider in the 'Tools' menu.
New icons in 'Misc' panel:
Map view toggle: [Optional] toggles display of the electron density map (if loaded). (See Tools sections to see how to load a map)
Map Contour toggle: [Optional] Contour density map around selected atoms. Contouring level can be adjusted in the Tools menu.
iMolview v1.2 Android
Load and contour electron density maps.
Tools/Measurement - This set of tools allows to measure distances, planar angles and torsions on displayed atoms. Tap on the tool you need and then sequentially select two,three or four atoms (depending on
the measurement type).
April 19th 2013: iMolview v1.1 for Android Released
A new version of iMolview for Android has been released.
The key new features are:
Release Version 1.1
Added molecular surface (skin) support
Added "Color Skin By Contact"
Added "Select Neighbors" button
Added "Select All" button
Option to color text sequence by 3D
Added Filter Selection by various properties
Added 'off line' information about PDB entry (References/Quick Info). (Note, that works only for newly loaded PDB objects)
April 6th 2013: Next generation of ICM desktop chemistry and biology modeling tools Workshop 2013
Thank you very much to all the participants of our workshop entitled "April 6th 2013: Next generation of ICM desktop chemistry and biology modeling tools Workshop 2013". It was an exciting workshop
highlighting all the new features of ICM and a half day user group session. Please click here to view the contents of the meeting and the speaker abstracts.
2012
November 11th 2012: iMolview v1.6 Released
A new iMolview v1.6 has been released and is available on iPad. For more information about iMolview please see:
http://molsoft.com/iMolview.html
The new features include:
Added interface to Dropbox (Documents/Dropbox)
Added molecular skin/surface representation
Added "Tools/Filter Selection" to adjust the current selection by various parameters
"Color Selection" can color individual representations.
ICM scripts embedded with the ICB file are available in the menu.
Added full screen mode.
Added interface to the ICM command language (Command tab in the search bar)
Tools menu (sbs stereo, measurements) appears for small molecules
2D labels are displayed properly
October 5th 2012: MolSoft Releases an Android version of iMolView
MolSoft has released its popular iMolview mobile software for the Android today. Please download iMolview from the Android version from the Google Play Store..
About iMolview Explore the fascinating world of biological macromolecules:
iMolview lets you browse and view in 3D protein and DNA structures from Protein Data Bank. Search for proteins like 'insulin' or 'thyroid receptor' in PDB or simply type pdb code the search bar. Information associated with each molecule in these databases is also at your fingertips. Sync and view your own structure files via iMolview folder on your sdcard. Molecular view can be customized with a rich set of molecular representations (wires, balls-and-sticks, space filling, ribbon diagrams, molecular surfaces) and various coloring schemes. Select residues, atoms or chains and color or change their representations individually. Select 'neighbors' of a ligand or any other selection to identify interacting atoms or residues. Set 'inertia' to the maximum and let your molecule spin in 3D indefinitely.
June 25th 2012: Fragment Based Drug Design using the 3D Ligand Editor
The ICM 3D Fully Interactive 3D Ligand Editor now contains tool which allows finding best scaffold replacement or linker fragment. It lets you interactively select two or more points of connection or replacement and search for possible fragments in the 3D database (SDF or Molsoft local database MOLT). The cutting bonds used to define scaffold are matched against a 3D database with a certain RMSD cutoff (usually very small). The best superposition is taken for the original compound and the chemical is minimized.
The result is returned as a .standard. Ligand Editor hit list where results can be easily browsed, sorted and compared. Linker prediction can be made in the presence or absence of the receptor. Filters such as energy strain, maximum number of rotatable bonds and contact distance to the receptor can be used. More documentation for this tool can be found here.
June 7th 2012: iMolview Version 1.5 Released
iMolview is an app for the iPhone/iPad that lets you browse protein, DNA, chemicals, and molecular documents in 3D. We have released a new version of iMolview (v1.5), which includes support for high resolution retina display, simultaneous display of multiple proteins and superposition, side-by-side stereo, and tools for measuring and displaying distances and angles. The app costs 99 cents and can be downloaded from iTunes. Please click here for more information.
June 5th 2012: New version of ActiveICM released.
A new ActiveICM download is available, please click here. We recommend that all users upgrade to the latest version. The new version fixes a bug that reported the error message .[588] too many different atom radii problem.. Please download the latest version now.
ActiveICM is a free plugin that enables you to view and display 3D ICM graphical slides and animations interactively inside Microsoft PowerPoint and web browsers such as Internet Explorer, Safari and Mozilla Firefox. The files can be prepared and saved in ICM Browser, ICM Browser Pro or ICM Pro. ActiveICM is being used for scientific publishing by journals such as PLoS ONE and Nature.
May 9th 2012: Impressive ICM Docking and Screening Benchmark Published
Last year scientists at MolSoft (Max Totrov Ph.D.) and UCSD (Ruben Abagyan Ph.D. and Marco Neves Ph.D.) competed in an industry-wide docking and screening competition organized by OpenEye, Merck, and GSK. Two challenges were set:
Pose Prediction
Docking, extended Astex set: 85 targets and 170 docking poses
Initial ligand conformations provided
Virtual Screening Recognition
VLS recognition: the DUD set (Useful Decoys)
40 targets, 10,000 decoys for each
MolSoft ICM achieved excellent results in both challenges ranking in first place in both challenges. ICM-Pro docking gave pose prediction results of < 1A top 1 pose: 78% of cases and < 2A top 1 pose: 91% of cases. For virtual screening, median area under ROC curves equal to 69% and 82% were achieved. A full description of the data analysis can be read here
April 4th 2012: User Group Meeting 2012 . Thank you to everyone who participated!
Everyone at MolSoft would like to thank all those who attended the ICM User Group Meeting this year. Thanks to all the great speakers for their excellent presentations. We had an interesting and enjoyable time which included talks on a wide range of topics including: Protein Structure Analyis, Membrane Protein Modeling, Modeling Immune Response Proteins, Latest ICM Developments, Chemical Biology and Cheminformatics, ICM for Teaching and Communication, Protein-Protein Docking, and Lead Discovery. Click on the links below to see what we all got up to - it was not all just science!
MolSoft is excited to announce the release of ICM version 3.7-2c. The new version can be downloaded from our support site here.
The key new features are described below but for a detailed list please see the release notes.
Atomic Property Fields APF is a 3D pharmacophoric potential implemented on a grid (Totrov 2008 and 2011 ). APF can be generated from one or multiple ligands and seven properties are assigned from empiric physico-chemical components (hydrogen bond donors, acceptors, Sp2 hybridization, lipophilicity, size, electropositive/negative and charge). An independent study found that APF is most accurate method for chemical superposition and ligand-based 3D virtual screening compared to several other commercial and academic softwares. (Giganti et al 2010).
Superimpose ligand binding sites by Atomic Property Fields [more..]
3D ligand Editor The ligand editor is a powerful tool for the interactive design of new lead compounds in 3D. It allows you to make modifications to the ligand and see the affect of the modification on the ligand binding energy and interaction with the receptor.
Multiple Receptor Docking in the 3D ligand editor [more..]
Chemical fragment linking in the 3D ligand editor [more..]
Dock to APF fields in the 3D ligand editor [more..]
Protein Modeling Inside ICM there are many features for homology modeling and loop modeling. This new option can be used if you have a gap in your protein and you want to find loops in the PDB which fit the gap.
New protein loop design method is now available. [more..]
Chemisty A couple of new options in the Chemistry menu.
Ligand-Based Screening Using Atomic Property Fields (APF)
Multiple Receptor Docking in the 3D Ligand Editor
Atomic Property Fields Docking in the 3D Ligand Editor
Virtual Ligand Screening (VLS) Result Analysis Tools
New VLS Hitlist Visualization Options
New High Precision Loop Modeling Tutorial
Details about our 2012 ICM User Group Meeting in March
2011
September 13th 2011: New iMolview Version Released - Now with Slides!
Today MolSoft released a new version (v1.4) of its popular molecular viewing app called iMolview for the iPad and iPhone. The new features include the ability to save molecular documents which include structure and slides. A slide is a combination of a view point and graphics representation. In iMolview you can create multiple slides and access them later after the document was saved.
To create a slide:
Setup a view and representations (color, label, etc.)
Go to Menu/Slides and choose "Save Current View"
Give a name and press 'OK'. It should appear in the list
Repeat these steps to create more slides
To switch between slides you may use Menu/Slides or arrows on the sides of the center button. After all slides are done you need to save your molecular document. Goto Menu/Documents/Save Current Document and give it a name. The document will be saved into History folder and can be loaded later. This file can also be copied to your PC,MAC or Linux and opened with free ICM-Browser, or you can embed this document as a live 3D object into web page using ActiveICM technology.
iMolview costs only 99cents and is available via the Apple App Store. Please send us your feedback on the new version and rate the app at the Apple app store.
A couple of years ago scientists using MolSoft's ICM software predicted and published the agonist bound structures of beta-adrenergic and adenosine A(2A) GPCR receptors. The accuracy of these models were confirmed by recently published x-ray crystal structures of these GPCRs. This work is summarized in a recent paper in Trends in Pharmacological Sciences. This work highlights the power of MolSoft's ICM modeling softare for the development of new ligands for GPCRs and also the use of these models for other pharmacological and biochemical studies.
September 1st 2011: Explore the Pocketome using ActiveICM
Prof. Abagyan's lab at UCSD have developed a website called the Pocketome which is an encyclopedia of conformational ensembles of all druggable binding sites that can be identified experimentally from co-crystal structures in the Protein Data Bank. The pocketome ensembles are generated from UniProt and PDB and clusters and validates the binding pockets based on the consistency of the composition between multiple structures. The pockets can then be viewed using ActiveICM or downloaded and viewed using ICM-Browser.
SummaryThe community-wide GPCR Dock assessment is conducted to evaluate the status of molecular modeling and ligand docking for human G protein-coupled receptors. The present round of the assessment was based on the recent structures of dopamine D3 and CXCR4 chemokine receptors bound to small molecule antagonists and CXCR4 with a synthetic cyclopeptide. Thirty-five groups submitted their receptor-ligand complex structure predictions prior to the release of the crystallographic coordinates. With closely related homology modeling templates, as for dopamine D3 receptor, and with incorporation of biochemical and QSAR data, modern computational techniques predicted complex details with accuracy approaching experimental. In contrast, CXCR4 complexes that had less-characterized interactions and only distant homology to the known GPCR structures still remained very challenging. The assessment results provide guidance for modeling and crystallographic communities in method development and target selection for further expansion of the structural coverage of the GPCR universe.
July 7th 2011: New Method to Visualize Protein Mutations Using ActiveICM
This month the Journal of Inherited Metablic Disease published a fully interactive 3D research paper using MolSoft's ActiveICM. The paper describes the crystal structure of human fumarate hydratase along with mutations that are mapped onto the 3D structure. The reader can click on a mutation or group of mutations and the fully interactive 3D display takes the reader to the location in the crystal structure in 3D. The reader can then interact (zoom, rotate etc..) with the molecule making the it easier to visualize the data. You can read more on the SGC website here.
June 17th 2011: iMolview gets reviewed on popular crystallography blog and store website P212121.com
We were excited to read a review of MolSoft's iMolview app for iPad and iPhone on the popular crystallography P212121.com website. Click here to read it... The P212121.com website is not only a blog but also a macromolecular crystallography equipment and reagent store. Scientists can browse thousands of products from dozens of companies all from a single website, this not only saves you time but you also avoid the sales reps! The iMolview app is being used around the world by scientists in many disciplines and we have received many positive reviews. The app is under active development, so if you are a user and there is a feature you would like to see please contact us. If you have not tried iMoview yet you can do so by purchasing it from the apple store for only 99 cents.
May 17th 2011: iMolview Version 1.2 Released
New in Version 1.2:
issue with older iPhone/iPod devices FIXED
structure files (pdb/icb) sync via iTunes
download history
easy toggle of individual molecule display via sequence view tabs
faster refresh in CPK representation
display tools popup bar on selection
improved handling of very large structures
neighbors in other chains selection, such as atoms in the vicinity of selected ligand
external links menu: Uniprot and RCSB entries in addition to PubMed
atom type coloring for hetero-atoms only - allows combining chain-specific colors and atom type color coding
3D residue picking is now tracked in sequence view
easy clear display button (X)
bug/crashes fixes
May 5th 2011: MolSoft releases iMolview for the iPhone and iPad
iMolview is an app for the iPhone and iPad that lets you browse protein, DNA, and drug molecules in 3D. The app has a direct link to the Protein Data Bank (PDB) and DrugBank and has a fast and easy to use interface. Touching the molecules via the screen allows you to interact immediately with the 3D structures in a unique way. You can zoom in and out, rotate, spin, pan, and clip the 3D molecules with your finger tips in ways that are impossible using a traditional mouse and desktop computer.
iMolView allows you to search for drugs such as 'ibuprofen' or 'gefitinib' in DrugBank, or proteins like 'insulin' or 'thyroid receptor' in the PDB and immediately explore their structure and annotation. This handy app allows you to quickly and easily view and interact with 3D molecules anywhere and at any time whether you are at the bench, at a conference, or just having coffee with colleagues!
May 5th 2011: Watch a Lecture by MolSoft's Principal Scientist Max Totrov Ph.D. about Ligand Binding Site Superposition and Comparison based on Atomic Property Fields
Max Totrov (Principal Scientist, MolSoft LLC) recently gave a lecture in Korea on "Ligand Binding Site Superposition and Comparison based on Atomic Property Fields". You can watch the lecture here and read more about this in this published paper.
February 2nd 2011: MolSoft awarded a patent for its ActiveICM Technology
MolSoft LLC were today awarded a patent entitled "Structured documents and systems, methods and computer programs for creating, producing and displaying three dimensional objects and other related information in those structured documents" for its ActievICM technology (US Patent Number 7,880,738). The patent is for a structured document file which includes graphical and non graphical information. The graphical information includes the vector and coordinate description of a representation of at least one three dimensional object and at least one of the following group of view attributes or features: rotation, translation, view angle, lighting, colors, specific graphical representations, labels and parameters. The vector and coordinate description are stored with or independently from the one or more view attributes or features and the vector and coordinate description and the one or more view attributes are stored in a single file which can be accessed and viewed by a structured document viewer. Related authoring tools and viewers are also disclosed.
February 1st 2011: First paper published in Nature Structural and Molecular Biology using ActiveICM
Nature Structural and Molecular Biology is now using using MolSoft's ActiveICM technology. The first paper published in this enhanced format in Nature Structural and Molecular Biology can be viewed here. Thank you to the Editors at Nature, the authors of the paper, and Dr. Brian Marsden and Dr. Wen Hwa Lee at SGC for their support and for making this possible. ActiveICM is developed by MolSoft and allows you to view fully-interactive 3D molecules and animations on the web or PowerPoint.
Dr. Ruben Abagyan who is Molsoft's founder and Professor at the Skaggs School of Pharmacy at UC San Diego said, "It is great to see ICM and its derivative, activeICM, revolutionizing the way scientific information is communicated to the public. Since their creation 20 and 6 years ago respectively we managed to transform this software into a sophisticated multi-platform environment. We are committed to the continuous development of this platform and making it freely available for the research community."
To view the ActiveICM content:
Display the HTML version of the paper in a web browser.
Download the ActiveICM plugin here or click on the link provided by Nature Structure and Molecular Biology (Image 1 and 2 below). If you click on the Enhanced Article link in the paper this will give you information on how to download the ActiveICM plugin..
Click on the hyperlinks in the html version of the paper and a popup ActiveICM window will be displayed (Image 3 below).
Use your left mouse button to rotate the molecule(s), zoom in by clicking on the left hand side of the display (Image 4 below).
Click on the MolSoft logo (bottom right corner) for more options.
January 12th 2011: MolSoft develops ChemDiv's Online Chemical Store
MolSoft recently developed ChemDiv's new eShop containing the world's largest small molecule inventory. The eShopuses MolSoft's proprietary cheminformatics tools to enable you to search the database by sketching the structure or part of a structure, by ID number, SMILES, or by chemical files such as Mol and SDF. A template structure can also be used to build libraries based around a particular substructure. There are also chemical property filtering tools available such as MW, logP, and Number of rotatable bonds, hydrogen bond donors and acceptors.
January 4th 2011: MolSoft Releases Major New ICM Version (v.3.7)
MolSoft is excited to announce the release of ICM v3.7-2 which is a major new version upgrade with many new features and an updated user interface. You can download the latest version at our support site. Click here for more information on the new features available in ICM v 3.7.
New feature hightlights include:
New ICM force field.
New easy to use HTML editor for creating molecular documents.
WebKit integration into ICM.
Charge protonation state prediction using a pKa model.
November 15th 2010: MolSoft Scientists Develop New ICM Force Field
Scientists at MolSoft have developed a new physics-based internal coordinate mechanices force field. Please see the publication in the Proteins journal by Arnautova et al where the force-field has been applied to protein loop modeling. The new force-field contains new parametization for the dielectric constant, an improved hydrogen bond determination method, and implementation of novel backbone atom torsional potentials which include bond anlges of the carbon (alpha) atoms into the internal variable set.
Follow these instructions to use the new force field in your own scripts for energy minimization and optimization. If you are interested in modeling loops you can use the _loopmodel macro where all these parameters are already pre-set.
Use ICM version 3.7-2a. This can be downloaded by logging into our support site.
The polypeptide must be built or converted with the icmff residue library to do this:
LIBRARY.res = {"icmff"}
readlibrary
The dielectric constant should be set to 2.
dielConst = 2.
In addition to regular energy terms, "bb" must be turned on
setterm"bb"
The force-field method should be set to icmff
ffMethod = 4
October 29th 2010: MolSoft LLC has moved office location.
After more than 10 years at Torrey Pines Court in La Jolla, MolSoft has moved to larger custom designed offices in Sorrento Valley. From the 1st November our new address will be:
11199 Sorrento Valley Road, S.209,
San Diego CA 92121
September 7th 2010: Dr. Brian Marsden (SGC Oxford) to Present iSee at Bio-IT World Conference
The development of ActiveICM otherwise known as iSee has been a result of a strong collaboration between
scientists at the SGC and developers at MolSoft. If you are attending the Bio-IT World Conference in Hannover next month please be sure to attend the talk by Dr.
Brian Marsden from the SGC entitled "iSee - Dissemination of Structural Biology Data to the Masses". The talk will be at 11:30AM on Wednesday 6th October in the IT Infrastructure and Informatics
session,please click here to read the abstract for his talk.
August 1st 2010: Molecular and Cellular Proteomics Journal Now Accepting Publications Using MolSoft's ActiveICM Technology
Following on from the success PLoS ONE and PLoS Biology have had using ActiveICM it gives us great pleasure to let you know that the Molecular and Cellular Proteomics Journal, published by the American Society for
Biochemistry and Molecular Biology (ASBMB) are now accepting articles in ActiveICM format.
Two articles have been published in this format already. Please click on the link below to view them. Remember to download the free ActiveICM plugin to view them.
Keren Lasker, Jeremy L. Phillips, Daniel Russel, Javier Velazquez-Muriel, Dina Schneidman-Duhovny, Elina Tjioe, Ben Webb, Avner Schlessinger and Andrej Sali (2010) Integrative Structure Modeling of Macromolecular Assemblies
from Proteomics Data August 1, 2010 Molecular & Cellular Proteomics,9,1689-1702. doi: 10.1074/mcp.R110.000067
July 29th 2010: MolSoft's Fully Interactive 3D Molecule Technology is Now Being Used by the PLoS Biology Journal
Click to enlarge image.
This week PLoS Biology have started using MolSoft's ActiveICM software to publish fully interactive 3D papers. This is following on from the recent success of the series in PLoS ONE. The articles allow the user to rotate, translate, and zoom 3D structures whilst reading the scientific paper and view animations directly linked to hyperlinked text. The publication of these papers is due to a collaboration between MolSoft and the SGC. Click on the links below and then choose Enhanced Version to view the latest papers in this fully interactive 3D format. You will need to download ActiveICM to view them.
Rellos P, Pike ACW, Niesen FH, Salah E, Lee WH, et al. (2010) Structure of the CaMKIIG/Calmodulin Complex Reveals the Molecular Mechanism of CaMKII Kinase Activation.PLoS Biol 8(7): e1000426. doi:10.1371/journal.pbio.1000426
Davis TL, Walker JR, Campagna-Slater V, Finerty PJ Jr, Paramanathan R, et al. (2010) Structural and Biochemical Characterization of the Human Cyclophilin Family of Peptidyl-Prolyl I
somerases.PLoS Biol 8(7): e1000439. doi:10.1371/journal.pbio.1000439
July 26th 2010: ICM-Pro Peptide Design Success Story: Design of a V3 HIV-1 gp120 mimotope.
In the current issue of Nature Structural and Molecular Biology scientists at New York University and MolSoft have reported the identification of conserved structural elements in the V3 crown of HIV-1 gp120 which could serve as epitopes to be targeted by a vaccine against HIV-1. A cyclic V3 mimotope peptide was designed using MolSoft's ICM-Pro by inserting a disulfide bond and four residue linkers between the preserved N and C terminals of V3. The peptide was designed by searching the Protein Databank for polypeptide segments that would match the ends of the V3 strands and the stability and positioning of the disulfide bridge were selected by energy minimization. The resulting mimotope was shown to interact with mAbs 1006, 2219 and 2557. The predicted structure and interactions of the mimotope were confirmed by X-Ray crystallography. This mimotope can potentially be used to design immunogens that could induce antibodies to V3 and other variable regions of gp120.
June 17th 2010: MolSoft's ICM Software Excels in Published Virtual Ligand Screening Comparison
In a recent publication by Giganti et al (Pasteur Institute, France) in the Journal of Chemical Information and Modeling ICM displayed the best performance in terms of molecular alignment and docking accuracy compared to four other software programs. The authors acknowledge the efficiency of MolSoft's Biased Probability Monte Carlo method which is good at sampling flexibility. You can read about the comparison here. The authors tested ICM-docking and the Atomic Property Field (APF) method. The APF method is described here: Totrov M (2008) Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem Biol Drug Des 71: 15-27
May 28th 2010: MolSoft's Virtual Screening Software Discovers New Potent Selective Inhibitor for p300 Histone Acetyltransferase
Click to enlarge image.
A collaboration between scientists at MolSoft and the John Hopkins University has resulted in the discovery of a potent selective small molecule inhibitor of p300/CBP Histone Acetyltransferase (P300 HAT). MolSoft scientists screened ~500K molecules using ICM-VLS and selected 194 for testing which resulted in 30 active compounds. P300 HAT is a transciptional activator implicated in many gene regulatory pathways and protein acetylation and is an important anticancer target. Read more here in the article published in Chemistry and Biology.
April 30th 2010:MolSoft's ICM Molecular Graphics in New Crystallography Textbook
MolSoft's ICM-Browser-Pro software was used to make all the molecular graphics images in Dr. Bernhard Rupp's new book called "Biomolecular Crystallography: Principles, Practice, and Applications to Structural Biology" published by Garland Science. To find out more about the book please click here and to read a review of the book please see this article in Acta Crystallographica Section D.
March 31st 2010: MolSoft Awarded New NIH Grant for a Glycosylation Modeling System for Internal Coordinate Mechanics
MolSoft has been awarded an NIH grant to work on developing a Glycoprotein modeling system for internal coordinate mechanics. The 2 year project will develop an extension to the current force-field (ECEPP) and a library of common glycosylatting chemicals for the modeling and docking of glycan-binding proteins. Glycans are key molecules in the immune system, cancer progression and cell-cell recognition and the ability to model them accurately is important for structure based drug design. You can read more about the aims of the grant here.
March 3rd 2010: Recent ICM Publications.
Please see some recent ICM publications including identification of new G-Protein Coupled Receptor (GPCR) antagonists using ICM-VLS (Katritch et al J. Med Chem 2010) and the modeling of the interactions of a Class B GPCR with its ligand using experimental restraints (Dong et al 2010). Another interesting paper by Rueda et al describes methods for the selection of experimental protein conformations for virtual screening.
2009
12.23.2009: End of Year Update
Below we have some end of year ICM news and a round up of some of our 2009 success stories.
MolSoft in the Media
Last month the San Diego Union Tribune wrote an article about MolSoft - you can read the article online here. In July the American Chemical Society publication Chemical and Engineering News also reported on our ICM 3D Ligand Editor. There was also a lot of interest by blog writers and other media outlets about MolSoft's ActiveICM being used by PLoS ONE and SGC scientists for the publication of fully interactive 3D scientific papers. SGC scientists refer to ActiveICM as iSee and they have summarized all the media interest in this development here in the whatisisee wiki.
Upcoming Workshops
This year we held two sold out ICM workshops and an ICM User Group Meeting. The User Group meeting was a fun and scientifically rewarding experience. You can see some of the photos from the User Group Meeting here (Photos by Dr. Lars Brive - Goteburg).
Please join us in 2010 at one of our 2010 workshops including an ICM Scripting Workshop in January - places are filling up quickly so please register as soon as possible.
Jan 21st-22nd, 2010: ICM Scripting Workshop. La Jolla CA
Apr 29th-30th, 2010: ICM Workshop:Protein Structure and Drug Discovery. La Jolla CA.
Oct 7th-8th, 2010: ICM Workshop:Protein Structure and Drug Discovery. La Jolla CA.
As mentioned earlier one of the big events this year was the exciting use of ActiveICM in the PLoS ONE journal to display fully interactive 3D objects. For the first time fully interactive 3D molecules were embedded in a peer-reviewed collection of research papers due to a collaboration with scientists at the SGC in Oxford UK. You can see the full PLoS ONE collection on their website.
You can use ActiveICM for free and download it from here.
Lots of New Cheminformatics Features
During the past year the number of chemistry related features inlcuded in ICM has grown significantly. We now have two chemistry products ICM-Chemist and ICM-Chemist-Pro. ICM-Chemist is ideal for all 2D chemical operations including sketching, editing, clustering and database enumeration. ICM-Chemist-Pro includes all the tools in ICM-Chemist plus 3D options such as chemical superposition and our popular 3D ligand editor. If you would like to try these products please let us know and we can send you an evaluation license.
New ICM-Chemist Tutorials Including Videos
Please check out the new series of ICM-Chemist tutorials including online videos. If you have ICM-Chemist or ICM-Pro-VLS then you have all the features described in these tutorials. More tutorials in this format will be released early next year so we would welcome your feedback.
Holiday Reading! Some Selected 2009 ICM Success Stories
ICM-VLS identifies new small molecule inhibitor for the avian influenza virus.
10.20.2009: MolSoft's New Technology Brings Online Publications to Life in Fully Interactive 3D.
Scientific publishing is now going 3D. For the first time in online publishing history, the PLoS ONE journal is publishing original research papers containing fully annotated and interactive 3D objects. This 3D environment provides a "movie-like" experience but with a significant difference - the reader is in control of the scenes and each view is linked directly to the text. This is possible because of a new technology called ActiveICM developed by MolSoft LLC. ActiveICM is now being used for scientific publishing due to a collaboration with scientists at the SGC. Initially the technology is being applied to the life sciences for the 3D display of the atomic structures of molecules implicated in disease. ActiveICM can also be used for any online publication where a 3D view would enhance the subject matter.
Click here to see the PLoS ONE collection of papers. Click here to read the press release.
10.08.2009: New MAC version of MolSoft's ICM Desktop Modeling Software is Now Available
Please visit our support site to download the latest MAC version 3.7-1f of ICM-Browser, ICM-Browser-Pro, ICM-Chemist and ICM.
10.07.2009: MolCart Building Blocks Database for Combinatorial Chemistry has been Updated
Click to enlarge image.
The MolCart Building Block database has been updated. The database consists of chemical building blocks for combinatorial chemistry from 32 different chemical vendors and contains 4,408,865 non-redundant chemical fragments. The database contains a wide variety of scaffolds which can be used to generate libraries using Molsoft's combinatorial library tools contained in ICM-Chemist. ICM-Chemist contains a large selection of tools for chemical enumeration and decomposition. You can create or modify a Markush structure and enumerate a library on the fly by adding chemical fragments to defined "R-groups". You can also enumerate by chemical reaction and decompose a database into chemical fragments.
9.22.2009: A new 64-Bit Beta Version of ICM is now available
A new 64-bit Beta version of ICM is available. Please contact andyATmolsoft.com if you would like to try this version of ICM.
9.19.2009: ICM Graphics On Front Cover of New Book on Chemogenomics
Click to enlarge image.
ICM graphics is featured on the front cover of a new book edited by Edgar Jacoby (Novartis) on Chemogenomics. More information about the book can be found here. The image is of the contoured electron density of a ligand. Click here for instructions on how to contour electron density in ICM.
7.22.2009: Updated MolCart Compound Database
Click to enlarge image.
The Molcart Compound Screening Database has been updated. The database is a collection of compounds from 33 chemical vendors who let us know whenever a new set of compounds is added or removed from their database.The database currently contains 8,275,223 compounds and the total number of redundant compounds is 4,474,597. The latest changes to the database are documented here. One of the advantages of the MolCart Compound Screening Database is that we take great care to ensure the compounds that are listed are actually available for purchase. Other compound database collections list compounds that are virtual and in theory can be synthesized but they are expensive and are rarely available for quick purchase. If you are a MolCart customer please update your database collection by clicking here and download individual vendor databases or our non-redundant compilation. If you are interested in purchasing MolCart you can get a 2 week trial license by contacting us at info@molsoft.com.
6.25.2009 Press Release San Diego, CA: Interactive Drug Design with the ICM 3D Ligand Editor
Click to enlarge image.
MolSoft LLC (www.molsoft.com) is a leading developer of breakthrough technologies in computational biology and chemistry. MolSoft has officially released a new ground-breaking tool for the interactive, three-dimensional (3D) design and docking of new drug candidates. The ICM Interactive 3D Ligand Editor is an easy-to-use desktop application that allows a chemist to visualize a receptor-ligand complex and then apply their experience and intuition to explore, design and optimize new drug-like compounds. Recently, this tool was validated in a community-wide evaluation of modeling and docking software where ICM performed very successfully in predicting the pose of a ligand to a G-Protein Coupled Receptor (see Michino et al Nature Reviews Drug Discovery 8: 455-456).
A common scenario in drug design is that a lead compound binds reasonably well to a target protein and the ligand-receptor interactions are known, but the problem of how to improve the binding properties, synthetic feasibility, and metabolic-physicochemical properties of the ligand remains. This is where the power of the ICM 3D Ligand Editor comes in: it allows the chemist to see the ligand-receptor interactions clearly on the computer screen. Chemists can simply explore their new ideas with a click of a mouse in a fully interactive and intuitive 3D environment. The effect of a change to the ligand in terms of its interaction with the receptor is immediately displayed on the screen for the chemist's immediate evaluation.
The pharmaceutical industry is already a major user of MolSoft's ICM 3D Ligand Editor technology and industry feedback has enabled MolSoft's developers to build a tool suited for chemists working in drug discovery research. Dr. Eric Martin, Director of Computational Chemistry at the Novartis Institute for Biomedical Research says "The ICM ligand editor is an extremely effective drug design tool for interactively merging the rigor of complex molecular calculations with the intuition of experienced medicinal chemists. We use it extensively both 1-on-1 at the graphics workstation, and in team meetings using a 3D projection system".
So how do you use the ICM 3D Ligand Editor? The process is simple and designed to make life easier for the chemist. First, the ICM 3D Ligand Editor displays the crystal structure or model of the receptor-ligand complex along with the properties of the pocket, ligand solvent accessible surface, ligand strain or unsatisfied hydrogen bonds. The chemist can then start exploring ideas for improving the ligand by selecting a new chemical group and adding it to the ligand. The binding energy and interaction with the receptor is immediately assessed and displayed on the screen. The chemist has access to a full panel of chemical groups, linkers, bond types, stereo/isomer options and atom selections, which can be used to quickly try many different ideas. Importantly, should something be undesirable, a key feature allows the chemist to quickly undo changes to the ligand with a single mouse click and get back to the original ligand. Once the chemist has designed a ligand they are happy with, it can be saved in a single file to share with colleagues. The editor is a perfect tool for communication among scientists and can aid brain-storming session in an exciting way because hypotheses can be tested instantaneously.
The ligand editor has many advanced functions that make use of MolSoft's much acclaimed ICM technology, which is the backbone of the tool. Inside the editor, features such as ligand minimization, docking, atom tethering and virtual screening are available to use. For example, thousands of chemical groups can be screened on a particular point of the ligand resulting in a ranked list of complexes. The editor can also be used for fragment based drug design and optimization. According to Professor Ruben Abagyan, MolSoft Founder and professor at the Scripps Research Institute, "This new interactive environment for lead discovery combines the best practices in flexible docking and interactive modeling. The user-friendly environment changes the way chemical probes are discovered in industry and academia. Furthermore, the freely available ICM Browser and ActiveICM plugin allow scientists to disseminate data among chemists and biologists in an interactive way."
The ICM 3D Ligand Editor is part of the ICM-Chemist-Pro software product available from MolSoft LLC (www.molsoft.com).
6.4.2009 New Feature: Display Alignment Profile
Click to enlarge image.
A popular request from the biologists who use ICM is to develop a way to visualize sequence conservation in an alignment. Therefore in ICM version 3.6-1e (to be released soon) we have developed a way to display the sequence profile in a multiple sequence alignment. The consensus strength of a residue at a point in the alignment is visually represented by the size of the letter in the profile (see accompanying image).
Click on the icon in the alignment to display the alignment options tool panel.
Check the profile option.
5.29.2009 MolSoft's ICM delivers star performance in GPCR modeling and docking competition.
Modeling structures of G Protein Coupled Receptors (GPCRs), the most prevalent drug targets, has always been a great challenge.
Predicting how and where the drugs bind to those receptors is an even greater challenge. Until recently only bovine rhodopsin and beta 2 adrenergic receptor structures have been solved. Yet, when the structure of the Adenosin receptor A2a (the receptor for caffeine) was solved in a complex with a drug, the crystallographers annouced a challenge for all modellers and dockers to predict the binding interactions of the drug with the receptor.
60 groups have registered for the competition and 29 dared to submit their models. Up to 10 models were allowed to be submitted. More importantly the models were supposed to be ranked since usually only the top predicted pose is further pursued.
In a recent paper by Michino et al published in Nature Reviews the results are published (click here to read the paper). Two teams from Molsoft, Katrich-Abagyan and Lam-Abagyan built the models that had the largest number of correct ligand-receptor interatomic contacts, 45 and 34 out of 70, respecitvely out of all participants. Moreover, both models were ranked number 1, while the prediction from a competing modeler thathad better drug RMSD (when a flexible drug ring was considered) was only ranked 2. The fourth model from the Baker/Rosetta group had only 18 of correct contacts and this pose was only ranked 4th.
The ICM-based GPCR modeling approach employs the Ligand-guided principle and the ICM global optimization algorithm, and its success in ligand docking shows applicability of the method to a large class of problems in the field of GPCR drug discovery.
5.15.2009 New Feature: Accent Graphics
A new edge accent feature is available in icm version 3.6-1e which will allow you to make images as shown in the pictures below.
The option can be activated in the command line using:
GRAPHICS.sketchAccents = yes
Click on the images below:
5.10.2009 Pictures from the 2009 ICM User Group Meeting
Picture by: Dr.Lars Brive (Goteborg University)
Thank you to everyone who attended the ICM User Group Meeting last month. It was a really enjoyable meeting with some great presentations. Click here to view the meeting program with speaker abstracts and bio.
Thank you to Dr. Lars Brive (Goteborg University) for his photos of the lunch at the beach (small/large) and also to Prof. Bengt Persson (Linkˆping University & Karolinska Institute) whose photos can be viewed here:
4.16.2009 Breaking News: Scientists identify compounds using MolSoft's ICM-VLS that might fight bird flu.
MolSoft's ICM-VLS was used by scientists in Canada and Hong Kong to screen the NCI ligand database to identify a synthetic compound which appears to be able to stop the replication of the influenza viruses, including bird flu virus. See the paper at J Med Chem webiste and here at MSNBC.
4.14.2009: MoSoft and Virginia Tech are awarded NIH grant to develop a new class of resistance-breaking insecticides to reduce malaria transmission.
Researchers from Virginia Tech and Molsoft LLC have received a five-year, $3.557 million grant from the National Institute of Allergy and Infectious Diseases (NIAID) to continue their promising work on a new class of resistance-breaking insecticides to reduce malaria transmission. Click here for the full story.
2.1.2009 ICM-Browser-Pro Image on Front Cover of Parasitology Journal
Thank you to Dr. Kandeel at Gifu University in Japan who let us know that one of the images from his paper in Parasitology was included on the front cover of the January issue of Parasitology. The image was made in ICM-Browser-Pro, see the front cover here.
2008
December 2008 Newsletter
MolSoft Newsletter: December 2008
In this issue:
End of Year Discount
Next ICM Workshop: Protein Structure and Drug Design February 5th to 6th 2009
2009 ICM User Group Meeting: Challenges in Protein Structure Prediction and Drug Design
New ICM Publication: Type-II Kinase Inhibitor Docking, Screening, and Profiling Using Modified Structures of Active Kinase States
Floating ICM licenses for the Mac are now available.
New Features in the latest ICM version 3.6-1d.
ICM Power Tip: How to search for chemicals in very large databases.
End of Year Discount
We are offering a 20% discount on all ICM-Pro and MolCart products. This special end of year offer applies to new customers or current customers who wish to add additional licenses to their existing license agreement. Submit your purchase order before December 31st 2008 and receive a additional 20% discount on all our ICM-Pro molecular modeling and cheminformatics products.
Next ICM Workshop: Protein Structure and Drug Design February 5th to 6th 2009
The workshop will be conducted by: Prof. Ruben Abagyan (The Scripps Research Institute) and Dr. Maxim Totrov (Principal Scientist, MolSoft LLC). The workshop will consist of lectures, demonstrations, and "hands-on" computational experiments and cover the following topics:
Sequence and Protein Structure Analysis
Protein Modeling and Simulations
Structure Validation and Optimization
Ligand Binding Site Prediction
Small Molecule Docking and Virtual Ligand Screening
Structure-based development of target-specific compound libraries
Cheminformatics, chemical clustering, searching, superposition ...
2009 ICM User Group Meeting: Challenges in Protein Structure Prediction and Drug Design
Please join us at our ICM User Group Meeting on April 9th and 10th to be held in La Jolla, CA. Come and hear about the latest developments in: Structure and Ligand Based Drug Design, Membrane Protein Modeling, Protein Structure Analysis, Virtual Ligand Screening, Protein-Protein Docking, Data Visualization, and Chemical Biology.
The registration fee for Academics is $99 and for Commercial is $300.
A registration form can be downloaded here.
New ICM Publication : Type-II Kinase Inhibitor Docking, Screening, and Profiling Using Modified Structures of Active Kinase States. Kufareva and Abagyan J. Med Chem
Abstract:Type-II kinase inhibitors represent a class of chemicals that trap their target kinases in an inactive, so-called DFG-out state, occupying a hydrophobic pocket adjacent to the ATP binding site. These compounds are often more specific than those that target active DFG-in kinase conformations. Unfortunately, the discovery of novel type-II scaffolds presents a considerable challenge, partially because the lack of compatible kinase structures makes structure-based methods inapplicable. We present a computational protocol for converting multiple available DFG-in structures of various kinases (?70% of mammalian structural kinome) into accurate and specific models of their type-II bound state. The models, described as deletion-of-loop Asp-Phe-Gly-in (DOLPHIN) kinase models, demonstrate exceptional performance in various inhibitor discovery applications, including compound pose prediction, screening, and in silico activity profiling. Given the abundance of the DFG-in structures, the presented approach opens possibilities for kinome-wide discovery of specific molecules targeting inactive kinase states.
Floating Licenses for the Mac
Due to customer demand we now offer floating licenses for the Mac platform. A floating license is placed on a server and any client computer connected to the server can ìcheck outî the license. In the past all ICM licenses for Mac were locked to a particular machine (nodelocked license). The floating license option now gives you more flexibility in your ICM use if you need it. You can receive a floating license for Mac the next time you renew or purchase a new license.
New Features in the Latest ICM Version
The new ICM developments are listed in the Release Notes here.
In particular please see:
Lots of new developments in the ICM Ligand Editor
New selections for Hbond donors and acceptors
New table coloring options
ICM Power Tip: Chemical searching very large databases.
ICM tables are capable of storing tens of thousands records. However, some problems operate with data sets so large that they do not fit in computer's memory. To work with such large amounts of data ICM uses the concept of Molsoft database (MOLT) files. Unlike many other table file formats, such as SDF, CSV and others, database files are optimized for fast search and other operations, like unique entry addition and diverse subset selection. To learn how to make a MOLT file and search see here.
November 2008 Newsletter
Welcome to the November 2008 MolSoft ICM Newsletter containing information on:
New ICM Cheminformatics Products Released: ICM Chemist and ICM Chemist Pro
Join us at the ICM User Group Meeting in April next year
2009 ICM Workshops - Protein Structure and Drug Design Dates
Google Search the ICM Manuals
See ActiveICM in Action
A New 3D File Format
ICM Method Publication: Atomic Property Field Superposition
Join the ICM Discussion Forum
ICM Power Tip: How to Make Molecular Movies in ICM
New ICM Cheminformatics Products Released
We are pleased to announce the release of two new stand alone Cheminformatics products called ICM Chemist and ICM Chemist Pro. ICM Chemist is a low cost feature-rich product for dealing with chemical structure and databases. The ICM Chemist Pro product contains all the features in ICM Chemist and gives you access to the 3D world of chemical viewing and manipuation. Some of the main features of each of the products are listed below
ICM Chemist Features
Read in and save chemical structures in a variety of formats.
Draw and edit chemical compounds.
Chemical Spreadsheets: Rich environment for display, manipulation and analysis of chemical datasets, from a few compounds to ~100K. For more than 100K you can use our MOLT file format (see below) and /or MolCart.
Chemical Searching
Fast herarchical clustering and trees.
Chemical Enumeration, Decomposition by R-Group (Markush Structures).
Build Local Databases (MOLT file): Large database files optimized for fast search and other operations such as unique entry addition and diverse subset selection.
Apply QSAR models.
and much more...
ICM Chemist Pro Features
Convert 2D chemical sketches to 3D and optimize.
Generate stereoisomers and tautomers.
Chemical superposition.
3D interactive ligand editor.
QSAR
Join us at the ICM User Group Meeting in April next year
April 9-10 2009 ICM User Group Meeting.
We are looking forward to the 1st ICM User Group Meeting in April next year. The meeting schedule will include talks by experienced ICM users from around the world with an emphasis on how they have successfully used ICM and other applications for their research. MolSoft's developers and scientists will also be presenting at the meeting, so it will offer a good opportunity for you to hear about the latest ICM developments and new features. More information about how to register for the user group meeting and the speakers will be in the next newsletter.
ICM Workshop - Protein Structure and Drug Design Dates
The dates for the 2009 ICM workshops to be held in La Jolla CA are listed below.The workshops will be presented by Prof. Ruben Abagyan (Scripps Research Institute, and Founder of MolSoft ) and Dr. Maxim Totrov (Principal Scientist, Molsoft).
Feb 5-6 2009 - ICM Workshop "Protein Structure and Drug Design"
October 1-2 2009 - ICM Workshop "Protein Structure and Drug Design"
The ICM Language Manual and the ICM User Guide Manual can now be google searched. Click here to give it a try. Once you have performed a search you can refine it to only display results from the Language Manual or the User Manual. This search will soon be integrated into the online versions of the manual.
See ActiveICM in Action
Click here to view an example of our new ActiveICM product (US Patent No:7,880,738) in action. The files used in ActiveICM can be generated using ICM Browser Pro, ICM Chemist, or ICM Pro and ActiveICM is free to customers of this product. The online example is a Structural Genomics Consortium datapack file of PAK4 kinase. Use your left mouse button to rotate the molecule, zoom in by clicking on the left hand side of the display, scroll down the text on the left hand side and click on the blue links to see different annotated views of the protein, and right click on the graphical display for more option, and right click on the graphical display for more options.
NOTE: The demo will only work if you download ActiveICM. Click here to download ActiveICM.
New 3D File Format
ICM now supports the 3D XML file format which is a popular 3D format for CAD. For example, Microsoft Virtual Earth uses this file format as well as CAD software companies such as Dassault Systems. There is a big online repository of 3DXML models here, models are free to download once you have registered. Once the file is downloaded, open the ICM graphical user interface, click on File/Open in ICM to bring the file into ICM and then display the file using the ICM Workspace.
New ICM Method Publication: Atomic Property Field Chemical Superposition
The Atomic Property Field (APF) superposition method is available in ICM Chemist Pro and the details of this method have been published this year by Dr. Maxim Totrov (Molsoft, Principal Scientist). Totrov M (2008) Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem Biol Drug Des 71: 15-27
APF is a 3D pharmacophoric potential implemented on a grid. APF can be generated from one or multiple ligands and seven properties are assigned from empiric physico-chemical components (hydrogen bond donors, acceptors, Sp2 hybridization, lipophilicity, size, electropositive/negative and charge). Click here to see how to use the APF method.
Join the ICM Online Discussion Forum
We now have over 80 members in the ICM Discussion Forum but this only represents a very small percentage of the ICM users worldwide. Click here to join the forum today. It is free to join the forum and there are many benefits of being part of this group: you will always be the first to get to know the new ICM features, you will learn new tips and tricks in ICM, find out how others are applying the software in their research, and most importantly you will be part of a network of people who can support and advise you when you have an ICM question. Click here to join the forum today.
ICM Power Tip
Making Molecular Movies in ICM
To make a movie in ICM all you have to do is click on the movie button located at the bottom of the graphical user interface. Any animation or view in the ICM graphical window will then be dumped into a movie file. For more instructions please click here.
07.11.2008 July 2008 MolSoft ICM Newsletter
MolSoft ICM Newsletter July 2008
Welcome to the July 2008 MolSoft ICM Newsletter containing information on:
All the exciting new features in ICM version 3.6 and links to the GUI manual.
A new product release - ActiveICM allows you to display ICM in PowerPoint and Web Browsers.
The next ICM Workshop in La Jolla CA October 2-3, 2008.
MolSoft will be at the Protein Society Meeting July 19-23, 2008
ICM Power Tip - Quick Ligand/Peptide Pocket Surface Display
New Features in the Latest ICM Release 3.6
Fully Interactive 3D Ligand Editor
The ligand editor is a powerful tool for the interactive design of new lead compounds in 3D. It allows you to make modifications to the ligand and see the affect of the modification on the ligand binding energy and interaction with the receptor. You can eidt the ligand by adding new functional groups, sampling a large collection of fragments on the fly, edit atoms or bonds, and and then in seconds see the affect of these changes on ligand binding. Clashes with the receptor are clearly flagged and energy contributions of each atom and total bidning energy and binding score are provided. Please see the graphical user interface manual for more information and instructions http://www.molsoft.com/gui/ligand-editor.html
You can now read, render and display any 3D object in ICM. e.g. KMZ and COLLADA format directly from the google sketchup website. We have also added options for changing the texture and material of a 3D object. See http://www.molsoft.com/gui/google-objects.html
Lots of New Data Plotting Features From ICM Tables
New plotting features include:
logarithmic scale
separate zooming for X and Y axis
zooming into/out of a certain point on the plot
Axis customization: setting titles and steps
Point labels in scatter plots, showing labels for selection only
Interactive control over histogram bin size to allow finding the best density estimation picture
Add any image you want to the background. This is great for making cool images or you can directly compare molecules by making a background image and then making changes to the molecule and see clearly the differences. See http://www.molsoft.com/gui/background.html
In many cases a compound database (sdf file) is so large that you cannot open it in ICM because you do not have enough memory on your computer. A way around this was to place the database on our product called MolCart but now you can build local chemical databases which you can browse and edit.
The occulusion shading option provides better representation of depth within a cavity. The color of each surface element of a grob (mesh) is changed by mixing its own color with the background depending on the burial of the surface element. See http://www.molsoft.com/gui/mesh-options.html#occlusion-shading
There is now a new layout for users that have a widescreen. Try it by going to File/Preferences/Gui Tab and change GUI.windowLayout
New Command Line Options
Other new command line options are described in the release notes .
New Product - ActiveICM
ActiveICM - Integrate ICM into PowerPoint or a Web Browser
ActiveICM will change the way you give presentations or share ICM data with your colleagues. With ActiveICM you can display a series of slides generated in ICM inside PowerPoint or a web browser. The slides are fully interactive which means the user can manipulate the molecules in 3D inside the viewer.
Click here to wownload ActiveICM and see the instructions here http://www.molsoft.com/gui/activeicm.html
Next ICM Workshop
The next ICM workshop will be held in La Jolla CA on October 2nd to 3rd. The workshop will be presented by Prof. Ruben Abagyan (Scripps Research Institute, and Founder of MolSoft ) and Dr. Maxim Totrov (Principal Scientist, Molsoft). See http://www.molsoft.com/training.html
Events - 2008 Protein Society Meeting
If you are attending the Protein Society 22nd Annual Symposium July 19-23, 2008 in San Diego be sure to stop by the MolSoft booth and say hello. We will be holding prize draws, demonstrating all the new ICM features, and we will be available to answer any ICM questions you may have.
ICM Power Tip
Quick Ligand and Peptide Pocket Display
Did you know you can quickly display the surface of a ligand or peptide by pocket by right clicking on the ligand or peptide in the ICM workspace and selecting Ligand Pocket. See http://www.molsoft.com/gui/dsPocket.html
06.20.2008 MolSoft @ Bio 2008
There were over 20,000 people at the Bio 2008 meeting and we thoroughly enjoyed demonstrating the latest ICM features and answering questions from the many people who stopped by at our booth. It was great to see so many ICM-users at the meeting and meet for the first time some of our international customers from Europe, Australia, and Japan. We received great feedback on our new ligand editor feature which will be released very soon, and our molecular document technology also attracted alot of interest. Other pictures from Bio2008 can be found here.
05.22.2008 MolSoft will be Exhibiting at Bio2008
MolSoft LLC is looking forward to presenting at the Bio2008 conference in San Diego June 17-20. Please come and visit us at booth #2539 where we will be showcasing for the first time lots of exciting new tools that will be released later this year in ICM Version 3.5-2.
04.08.08 New ICM Paper: New method for the assessment of all drug-like pockets across a structural genome.
Nicola G, Smith CA, Abagyan R. New method for the assessment of all
drug-like pockets across a structural genome. Comput Biol. 2008 Apr;
15(3):231-40.
Department of Molecular Biology, The Scripps Research Institute, La
Jolla,
California.
Abstract:
With the increasing wealth of structural information available for
human pathogens, it is now becoming possible to leverage that
information to aid in rational selection of targets for inhibitor
discovery. We present a methodology for assessing the drugability of
all small-molecule binding pockets in a pathogen. Our approach
incorporates accurate pocket identification, sequence conservation
with a similar organism, sequence conservation with the host, and
structure resolution. This novel method is applied to 21 structures
from the malarial parasite Plasmodium falciparum. Based on our survey
of the structural genome, we selected enoyl-acyl carrier protein reductase (ENR) as a promising candidate for virtual screening based inhibitor discovery.
03.20.08 New ICM Publication: A new method for ligand
docking to flexible receptors
New ICM publication by the Scripps Research Institute and MolSoft
scientists.
See:
Bottegoni G, Kufareva I, Totrov M, Abagyan R. A new method for ligand
docking to flexible receptors by dual alanine scanning and refinement
(SCARE). J Comput Aided Mol Des. 2008 Feb 14;
Abstract
Protein binding sites undergo ligand specific conformational changes
upon ligand binding. However, most docking protocols rely on a fixed
conformation of the receptor, or on the prior knowledge of multiple
conformations representing the variation of the pocket, or on a known
bounding box for the ligand. Here we described a general induced fit
docking protocol that requires only one initial pocket conformation
and identifies most of the correct ligand positions as the lowest
score. We expanded a previously used diverse "cross-docking" benchmark
to thirty ligand-protein pairs extracted from different crystal
structures. The algorithm systematically scans pairs of neighbouring
side chains, replaces them by alanines, and docks the ligand to each
'gapped' version of the pocket. All docked positions are scored,
refined with original side chains and flexible backbone and re-scored.
In the optimal version of the protocol pairs of residues were replaced
by alanines and only one best scoring conformation was selected from
each 'gapped' pocket for refinement. The optimal SCARE (SCan Alanines
and REfine) protocol identifies a near native conformation (under 2 A
RMSD) as the lowest rank for 80% of pairs if the docking bounding box
is defined by the predicted pocket envelope, and for as many as 90% of
the pairs if the bounding box is derived from the known answer with
approximately 5 A margin as used in most previous publications. The
presented fully automated algorithm takes about 2 h per pose of a
single processor time, requires only one pocket structure and no prior
knowledge about the binding site location. Furthermore, the results
for conformationally conserved pockets do not deteriorate due to
substantial increase of the pocket variability.
03.11.08 Free Online Tutorial Dates Released for May through to July.
Click here for information on the upcoming online demonstrations.
03.03.08 New Review Article from MolSoft on Flexible Docking
Here is a link to a new publication in Current Opinion Structural
Biology on flexible docking by Dr. Totrov (MolSoft) and Prof. Abagyan (Scripps). The paper highlights all the recent development for including multiple receptor conformations in docking to represent ligand induced fit.
02.14.08 New Tutorial: How to model the effect of a mutation on protein structure.
Click here for a new tutorial on how
to make a mutation in a protein structure and optimize.
02.14.08 MolSoft Scientists Collaborate with Schering Plough to Identify New GPCR Inhibitors using ICM.
Click here to read how MolSoft and Schering Plough scientists identified new inhibtitors for a GPCR using MolSoft's drug discovery products. The structure-based development of MCH-R1 and other GPCR antagonists is hampered by the lack of an available experimentally determined atomic structure. A ligand-steered homology modeling approach has been developed (where information about existing ligands is used explicitly to shape and optimize the binding site) followed by docking-based virtual screening. Top scoring compounds identified virtually were tested experimentally in an MCH-R1 competitive binding assay, and six novel chemotypes as low micromolar affinity antagonist "hits" were identified. This success rate is more than a 10-fold improvement over random high-throughput screening, which supports our ligand-steered method. Clearly, the ligand-steered homology modeling method reduces the uncertainty of structure modeling for difficult targets like GPCRs.
02.04.08 New Tutorial: How to dock to multiple receptor conformations.
The tutorial uses Aldose Reductase as an example. Aldose Reductase has
a flexible loop in the ligand binding pocket vicinity which enables a
variety of inhibitors to bind and therefore in order to identify these
ligands via docking it is necessary to sample the conformations of
this loop and dock to an ensemble of structures.
2007
12.15.07 New Docking tutorials - Docking to Electron Density and Focused Markush Library Docking
How to dock a ligand to an electron density map. The ICM X-Ray AutoFit is an automated method to fit a ligand into electron density. The tool combines the powerful ICM docking algorithm with an electron density fitting function. Click here for step by step instructions.
How to dock a focused Markush library. A Markush library can be generated on the fly and docked using ICM. In this example we use the roscovitine ligand bound to CDK5 to identify scaffolds which may improve ligand-receptor interaction.
10.23.08 New Slide Transition EffectIn ICM version 3.5-1l a new blending transition effect between slides
is available. To see this in action download the latest version of ICM
or the free ICM-Browser and view this icb file:
Make a couple of slides - click camera button at bottom of the gui
- Note: to see the blending transition the transition needs to be made
between different representations eg wire to ribbon
Right click on the name of the slide in the ICM workspace and
select edit slide
At the bottom of this window you will see options for the currently
available transitions - blend and smooth - check which one you would
like to use and the transition time in ms.
October 22nd 2007
As a quarter of million people fled their homes amid fierce wildfires that had burned 100,000 acres around San Diego County, Molsoft opened its doors for the families of the escapees. Children, pets, and adults have found a comfortable home at Molsoft and were given a place to stay, sleep, access internet and play video games.
Games, food, laughter and partial property destruction made their stay at Molsoft more comfortable. The training class became an information center from which the families were following the fate of their homes.
October 17th 2007
MolSoft launches the ICM Forum. Please sign up and contribute with questions/answers and interact with other ICM users worldwide.
October 12th 2007
MolSoft announces a series of online demonstrations as a new way of teaching the ICM suite of software. Click here for more information.
July 12th 2007
Molecular Movie Making Made Easy Real-time screen grabbing movie making is now available in ICMBrowserPro on all platforms. With one click of the mouse anything displayed in the graphical displaywindow will be transfered to movie format - mpeg, avi or mov. See an example of an ICM movie here.
June 2nd 2007
MolCart Compound Database Update
The MolCart Compound screening library has been updated. The database contains 3,621,728 unique compounds for use in MolCart. Please contact info@molsoft.com for more information on licensing MolCart.
May 25th 2007
The latest version 3.5 for all ICM products has been released for Windows, Mac , Linux and SGI.Please download the latest version of ICM from our support center.
Some of the new features include:
Screen-grabbing movie making - one click movies saved in mpeg, avi or mov format.
Multiple-receptor docking to represent flexibility within the ligand binding site.
Faster machine learning tools for QSAR.
Over 20 new chemistry menu options added.
Improved graphics and new styles for xstick, variables, transparent CPK, hydrogen bonding, chain gaps.
March 27th 2007 Maxim Totrov PhD to present data at American Chemical Society National Meeting
On March 27th 2007 Dr. Maxim Totrov (Principal Scientist - MolSoft LLC) will be giving a presentation entitled "Chemical superposition and pharmacophore elucidation by SCAPFOld: Self-consistent atomic property field optimization" at the American Chemical Society 233rd National Meeting & Exposition, Chicago, IL USA. The presentation abstract can be found here.
March 22nd to 23rd 2007 ICM Workshop
Once again the ICM Workshop sold out quickly and we had a full class of students eager to learn about Protein Structure and Drug Discovery using MolSoft's ICM software. The first day and a half of training was presented by Prof. Ruben Abagyan (Scripps Research Institute). Prof. Abagyan gave lectures and demonstrations on ICM sequence and structure analysis, molecular modeling and cheminformatics. Friday afternoon was devoted to small molecule docking, virtual ligand screening and protein-protein docking led by Dr. Maxim Totrov (Principal Scientist - MolSoft). The participants ran docking examples such as virtual screening of small ligand databases to identify known inhibitors of the cyclooxygenase receptor and protein-protein docking of subtilisin and chymotrypsin. All the participants agreed that the workshop will help them greatly when they return to the lab and would highly recommend the training to their colleagues.
If you were unable to attend this workshop we are holding workshops in May and September this year. Please click here for more details and sign up early to guarantee your place.
2006
August 1st 2006 MolSoft Awarded NIH SBIR Phase I Grant to develop protein surface annotation software
MolSoft was recently awarded a NIH SBIR Phase I Grant to develop an integrated protein surface annotation suite of software for biologists and chemists. The software will allow users to apply several binding site prediction methods and analyze the results. These new algorithms include a protein-protein interface prediction method for small molecule binding sites and a surface structural motif detection and visualization method.
July 1st 2006 MolSoft Receives Bill and Melinda Gates Foundation AIDS Research Grant
Scientists at MolSoft LLC will be working as part of a multidisciplinary team led by Susan Zolla-Pazner Ph.D. (New York University School of Medicine) on a grant funded by the Bill and Melinda Gates Foundation entitled "The V3 Loop: A Conserved Structure of gp120 that Can Induce Broadly Neutralizing Antibodies Against HIV-1"
MolSoft LLC is excited to be leading the structure-based engineering of the V3-carrier immunogen. Using our experience with similar efforts we will undertake ab initio loop modeling of protein segments consisting of the central portions of the V3 signature sequences and modeling of carrier joining sequences. For more information please see the press release in the
New York Times.
July 1st 2006 MolSoft and Virginia Tech collaborate to work on malaria vector control
MolSoft and Virginia Tech will be working together on a 3-year project funded by the Foundation of the National Institutes of Healt to apply state-of-the-art computer modeling and chemical synthetic approaches to produce highly potent and selective anticholinesterases (AChEs) for malaria vector control. MolSoft's ICM software will be used to build homology models of AChEs and virtual screening will be applied to identify selective inhibitors. ICM-Chemistry will then be applied to optimize the lead compounds.
February 8th 2006: From Physics to Biology: the interface between experiment and computation BIFI 2006 - II International Conference, Zaragoza Spain.
Claudio Cavasotto PhD (Senior Scientist - Molsoft) has been invited to present a talk entitled "Ligand docking and virtual screening in structure-based drug discovery" at the
BIFI 2006 - II International Conference, Zaragoza, Spain. His talk will be on February 8th 2006 and more details about the conference can be found
here.
January 25th-27th 2006 Three-Day ICM Workshop.
On January 25th-27th 2006, a 3-day ICM Workshop was held at Molsoft's La Jolla
offices. The first two days of the workshop covered all aspects of protein
structure and drug discovery including sequence analysis, protein modeling,
small molecule docking, protein-protein docking, cheminformatics and QSAR. The
third day was dedicated to advanced ICM training including the ICM command
language, scripting, loop modeling and flexible receptor docking. The course was conducted by Prof. Ruben Abagyan (The Scripps Research Institute) and
Dr. Maxim Totrov (Principal Scientist - Molsoft).
January 1st 2006 MolSoft and the Mayo Clinic College of Medicine Collaboration
Scientist at the Mayo Clinic and MolSoft will be collaborating on a project to model G-protein coupled receptors (GPCRs) which are key receptors for a large number of disease pathways. The aim is to use MolSoft's proprietary software to build highly-refined and rigorously validated models of GPCRs and to use these models to guide experimental work at the Mayo Clinic.
2005
September 29th-30th 2005 Successful Workshop
On September 29th-30th an international group of ICM users from academia and industry gathered for our ICM Workshop entitled "Protein Structure and Drug Discovery" Click here to see what went on.
August 3rd, 2005 New Version Release
Molsoft has released the latest version (3.4) of ICM. This version includes a number of new features as described in the Release Notes.
The most significant addition is the incorporation of ICM Molecular Documents and Presentation (US Patent No:7,880,738). These can be constructed and viewed in ICM, they
can consist of text linked to molecular animations and transitions. The animations and transitions are fully-interactive and can be interrupted without any loss
of information. For more information on ICM Molecular Documents and Presenations please click here.
April 14th, 2005 New Product Release. MolCart is the latest addition to the ICM suite of software. MolCart allows you to store and manipulate
large chemical databases which can then be searched and analyzed using ICM cheminformatics tools.
April 25th, 2005 New SGI version release. A new ICM SGI version - 3.3-04c has been released. Please click here
to download the latest version.
April 5th, 2005 The Latest version of ICM (3.3-04) has been released. Please click here
to download the latest version. Release Notes.
2004
August 10th, 2004 Press Release:San Diego, CA. Molsoft LLC and the Structural Genomics Consortium at the University of Oxford Announce Software Co-Development and Research Collaboration. more details
July 15th, 2004 Molsoft LLC is pleased to announce a collaboration with the Burnham Institute, La Jolla, on a Tobacco-Related Disease Research Program Funded Grant entitled
"Vitamin A Derivatives as Antagonists of Nicotine Effects".
July 30, 2004 California State University at Fullerton Expands Use of ICM-Pro to New Computational Biology Curriculum Being Offered in 2005-06
Molsoft is pleased to learn that the Department of Chemistry and Biochemistry at California State University, Fullerton, is expanding its teaching use of ICM-Pro.
CSU Fullerton began using ICM-Pro for training undergraduate chemistry and biochemistry students in Spring 2004.
ICM-Pro has also been used to teach structural bioinformatics to students seeking a Certificate in Bioinformatics through CSUFs Extended Education program.
Beginning in 2005-06, ICM-Pro will be featured in a new computational biology program for chemistry and biochemistry majors [more].
July 20-21st Molsoft Conducts ICM Workshop in New Jersey USA
On the 20-21st of July 2004 sixteen people attended the ICM workshop entitled "In Silico Drug Discovery".
The workshop was held at Bristol Myers Squibb at Princeton New Jersey.
June 25th, 2004
Molsoft LLC is pleased to announce a collaboration with the Burnham Institute, La Jolla, on a STTR NIH funded grant entitled
"15 - Deoxy -12,14-prostaglandin J2 as a ligand of RXRalpha".
March 17, 2004 Molsoft LLC Receives Phase I STTR to Develop Beta-Catenin Antagonists
Molsoft LLC received a Phase I STTR award from the National Institutes of Health, National Cancer Institute, for the project entitled.
Rational Development of TCF/Beta-Catenin Antagonists. The project will combine Molsoft's novel in silico lead development platform with the
cancer biology resources at The Burnham Institute to discover and characterize small molecule ligands using
the beta-catenin structure both alone and in complex with TCF. Beta-catenin signaling has been implicated in a number of malignancies,
which makes the beta-catenin/TCF interaction an promising target for the prevention and treatment of a variety of tumors and leukemia,
and the project proposes to develop a novel generation of drugs active against various forms of cancer.
2003
July 25, 2003 Molsoft LLC Receives Small Business Biodefense Grant Award
Molsoft LLC received a Small Business Biodefense Program award from the National Institutes of Health, National Institute of Allergy and Infectious
Diseases, for the project entitled .Rational Design of Inhibitors of Yersinia pestis EF-Tu..
Molsoft and its research partners at the University of California, Irvine and Chemical Diversity Labs, Inc., San Diego,
propose to develop a new class of antibacterial agents active against Category A pathogens likely to be used by bioterrorists
including those causing plague, anthrax, cholera, and typhoid fever.
Furthermore, the project proposes to develop design principles that will be useful in developing agents less susceptible to the problem of
rapidly emerging antibiotic drug resistance.
18 June 2003.Mpex Pharmaceuticals and Molsoft announce collaboration for the structure-based design of new antibacterial compounds.
Mpex Pharmaceuticals Inc. (ìMpexî) and Molsoft, LLC (ìMolsoftí) have announced a
collaboration to rationally design new antibacterial agents based on the
refinement of a 3-D molecular structure of a membrane protein target,
the functional characterization of its surfaces, and the application of
predictive algorithms ..more..
June 16, 2003 Molsoft LLC Receives Phase II STTR Grant Award to Continue Developing Thyroid Receptor Antagonists
Molsoft LLC received a Phase II STTR award from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases,
for continuation of the "Rational Development of Thyroid Receptor Antagonists. project. In Phase I, Molsoft and its collaborators at the
NYU School of Medicine and the NYU Department of Chemistry achieved proof of concept. Molsoft.s virtual ligand screening
technology was used to discover 14 small molecule thyroid receptor antagonists displaying extreme structural diversity with
IC50s ranging from 4 to 30 microM. Additionally, the test-case lead optimization scheme designed in Phase I based on generating focused
virtual libraries of molecules easily amenable to organic parallel synthesis resulted in the rapid identification of second generation hits
with IC50 in the nanomolar range. In Phase II, Molsoft and its NYU research partners propose to conduct full-scale optimization cycles
of selected hits identified in Phase 1 in order to produce low nanomolar hits and to evaluate those hits using computational tools,
in vitro characterization, and preliminary animal studies.
May 20th-21st 2003 Molsoft LLC Conducts Annual Spring Training CourseOn the 20-21st May 2003 conducted an ICM workshop entitled "In Silico Drug Discovery".
The workshop was held at Molsoft LLC La Jolla.
2002
21 October 2002. Molsoft to Evaluate Novel Scaffolds for PDF Inhibitor Program
Molsoft LLC, a La Jolla based company providing new, breakthrough technologies
in computational chemistry and biology, and GeneSoft Pharmaceuticals Inc., a
specialty pharmaceutical company headquartered in South San Francisco,
announced that they have signed a collaborative agreement to evaluate
inhibitors of peptide deformylase (PDF), an essential bacterial metalloenzyme
and novel antibiotic target ..more...
26 August 2002. Molsoft Conducts Training Course/ Workshop in La Jolla, CA
Molsoft conducted an intensive 2-day training course/workshop on the ICM suite
of products at La Jolla, CA. The lecture and hands-on demonstrations covered
topics of interest to computational chemists and biologists in the area of drug
discovery. The topics included molecular graphics, animation, bioinformatics,
homology, protein modeling, docking and ICM command language for expert users.
The attendance was international with scientists coming from Asia and Europe as
well as from various parts of the US. The attendees were interested in having
future courses, both at the basic and at the advanced levels.
One of the attendees from Australia who is very well known in the modeling
field for his early work in structural bioinformatics, Dr. Jiri Novotny
(Consultant for Biocomputing and Bioinformatics), said, "The course was well
organized and presented the participants with two full days of new and
efficient tools applicable to the most pressing problems of current structural
biology and bioinformatics: structural/functional annotation of genomic data,
analysis of protein surfaces, ligand binding sites, virtual ligand screening,
structure derivation, regularization and model building". He continued on to
say, "Perhaps the most impressive aspect of the workshop was the skill,
dedication and the highest professional standard of the scientific team
established at Molsoft by Prof. Abagyan. In hands-on session at computer
screens the participants were encouraged to ask questions and bring out
problems that were answered and solved immediately by the whole team including
themselves. It was an invaluable practice and a good demonstration of the speed
and versatility of the newest ICM version."
The new version, ICM 3.0, was introduced at the course and was well accepted as
an extremely user-friendly interface by the attendees. This version, which also
has stereo viewing mode that works with VRex glasses on Linux and Windows
platforms, was welcomed by all the participants as an efficient and low cost
alternative to existing stereo viewing tools.
About the training course, Dr. Maxim Totrov, co-founder and Principal Scientist
at Molsoft said, "The course provided us with a vital opportunity to meet
members of the growing community of ICM-users and bring them up-to date with
the latest developments in ICM. It was exciting to see first-time users perform
complex molecular modeling tasks such as protein-ligand docking after only a
few hours of training. These workshops are geared towards helping our customers
utilize fully the power of ICM molecular modeling suite in their everyday
work."
22 August 2002. Molsoft Participates in the Annual Symposium of the Protein Society in San Diego
Molsoft is pleased to announce its participation at the Protein Society's 16th
Annual Symposium held at the Marriott in San Diego from August 17-21, 2002.
Attendance was estimated at around 1300. Molsoft's exhibit was designed to
inform Protein Society attendees about the latest technology available from
Molsoft including its recent version of a new graphical interface which enables
Molsoft's ICM software easier to use than ever before.
"We enjoyed talking to numerous visitors to our booth," said Dr. Lalitha
Subramanian, Director of Contract Research for Molsoft, "many of whom were
extremely excited to see ICM's new GUI interface." She added, "Our science has
always been strong and well validated. Now, with the added power of a very user
friendly interface, ICM will be the choice for most researchers in the academic
and commercial fields."
Molsoft also participated in the Beyond Genome 2002 conference held in June of
this year and will be presenting at the BioITWorld conference to be held in
November.
Molsoft LLC Receives Phase II SBIR Grant Award May 21, 2002
Molsoft received a Phase II SBIR award from the Department of Energy for
continuation of the "GAP: Genomics Annotation Platform" project. In Phase I
Molsoft used its many years of experience in developing molecular visualization
and manipulation software to built the computational tools central to the
functionality of the GAP. In Phase I, Molsoft successfully used GAP to predict
the function of uncharacterized genes. In Phase II, Molsoft will optimize the
existing tools and develop an efficient graphical user interface while adding a
system for automatic updates to GAP. When completed, the Molsoft GAP will
provide an online research platform allowing users from the pharmaceutical
industry to access Molsoft's state-of-the-art computational biology technology
and to use that technology for extracting meaningful information from the human
genome project and from other major, international sequencing efforts.
March 2002
Molsoft received a Phase 1 SBIR grant from the National Institutes of Health,
National Human Genome Research Institute for its project entitled
"Sequence/Structure Annotation of Protein Families." The goal in this project
is analyzing in silico well known protein families in order to identify which
family members could be good therapeutic targets and how pharmacogenomics
strategies can be devised to better design drugs targets at these proteins.
2001
August 2001
Molsoft and Plexus Vaccine Inc.entered into a licensing agreement to discover
and commercialize new interventional strategies for global infectious diseases,
for drug-resistant microbes, and for chronic diseases associated with cryptic
pathogens. Plexus will use the Molsoft ICM software suite to develop Virtual
Epitope databases of molecular disease targets for in silico design of
structural mimics that can be readily synthesized, tested, and formulated as
vaccines ..more..
August 2001
Molsoft received a Phase 1 SBIR grant from the Department of Energy for its
project entitled "GAP: Genomics Annotation Platform." The goal in this project
is to develop bioinformatics tools used for predicting the function of
uncharacterized genes.
May 2001
Molsoft and HTS Biosystems, Inc. (HTS) entered into collaboration for Molsoft
to develop software modules for instrument control, data collection, local
visualization, and analysis of data generated by HTS' Surface Plasmon Resonance
(SPR) instruments.
April 2001
Molsoft LLC and Chemical Diversity Labs, Inc. (CDL), San Diego, CA, formed a
strategic alliance to provide joint chemistry and computational modeling
services to their customers. Molsoft will use known three-dimensional
structures of drug targets as the starting point for the rational selection
process. The Molsoft modeling by homology technology will be applied in cases
when the structure is not available. CDL will give Molsoft an access to its
collection of over 250,000 diverse purified small molecules already available
for biological screening.
February 2001
Molsoft and Biovitrum AB, a subsidiary of Pharmacia & Upjohn AB, entered into
an agreement to co-develop a Chem-Informatics Client System. Molsoft and
Biovitrum will use Molsoft's proprietary programming languages and libraries to
develop the software, which will be called BeeHive and will function as a
program manager and database interface for chemical searches, analysis, and
integration.
2000
November 2000
Molsoft has moved to a new location, click the press release for more details.
October 2000
Molsoft and Syrrx, Inc., a pioneer in the field of structural proteomics,
entered into a strategic alliance to accelerate structure-guided drug discovery
through the combination of Molsoft's Virtual Ligand Screening (VLS) technology
and other computational biology technologies with Syrrx's high-throughput
structural proteomics platform. ..more..
September 2000
Molsoft received a Phase 1 STTR grant from National Institutes of Health,
National Institute of Diabetes and Digestive and Kidney Diseases for its
project entitled "Rational Development of Thyroid Receptor Antagonists." The
goal of this project is to discover and optimize small molecule antagonists of
the thyroid hormone receptor. Molsoft will work with the New York University
School of Medicine to complete the project.
September 2000
Molsoft LLC announced the relocation of its headquarters and commercial
development operations to an expanded facility in La Jolla. The new facility
will accommodate several expanded programs, including the continued refinement
of ICM 2.8, the expansion of software sales and the addition of new products
and in-house services. The new facilities include 3500 square feet of computer
laboratory, research, training, and office space in close proximity to The
Scripps Research Institute and the Genomics Institute of the Novartis Research
Foundation (GNF), both of whom have research collaboration agreements with
Molsoft.
September 2000
Molsoft LLC announced its first shipment of the Molsoft BioPackage to Hitachi,
Ltd. for distribution in Japan. During the two-year Distribution Agreement,
which became effective June 1, 2000, Hitachi will be the exclusive distributor
of Molsoft's BioPackage in Japan. Hitachi plans to market the software to
Japanese corporations, universities, and laboratories. Molsoft's computer
software provides novel computational and informational technologies for
biomedicine including genomics and drug discovery. ..more..
April 2000
Molsoft and The Genomics Institute of the Novartis Research Foundation (GNF)
have entered into a Research Collaboration Agreement. The collaboration will
focus on the further development of computational biology tools for functional
and structural annotation of new genomic sequences, protein modeling, and
virtual ligand screening. Molsoft will provide its ICM software for research in
functional and structural annotation of new genomic sequences, protein modeling
and lead discovery at GNF, which will provide GNF researchers with an accurate,
speedy and flexible method for structural annotation and protein structure
modeling. Molsoft will grant GNF a nonexclusive license to use Molsoft
databases and software in gene discovery, functional genomics, and drug
discovery, and GNF will supply support for personnel and computer facilities to
be used in the joint tool development projects ..more..
July 2000
Molsoft LLC and The Scripps Research Institute (TSRI), a private, non-profit
research organization engaged in basic biomedical research, announced the
formation of research collaboration. During the term of the agreement, TSRI and
Molsoft will collaborate on defined joint projects involving the functional and
structural annotation of new genomic sequences and protein modeling. For the
joint projects, Molsoft will grant TSRI a nonexclusive and restricted license
to use Molsoft databases and software in gene discovery, functional genomics
and drug discovery, and TSRI will supply personnel and computer facilities for
use in the joint projects. ..more..
1999
November 1999
Molsoft LLC and eBioinformatics, Inc. announced the formation of an alliance in
which Molsoft will provide its ICM software for incorporation into
eBioinformatics, Inc. flagship product BioNavigator. The Molsoft-ICM suite of
computational biology tools will provide BioNavigator users with access to the
latest and most powerful algorithms. This alliance will give scientists new
opportunities based on the power of Molsoft's ICM software and the convenience
of BioNavigator's web-based bioinformatics workspace ..more..


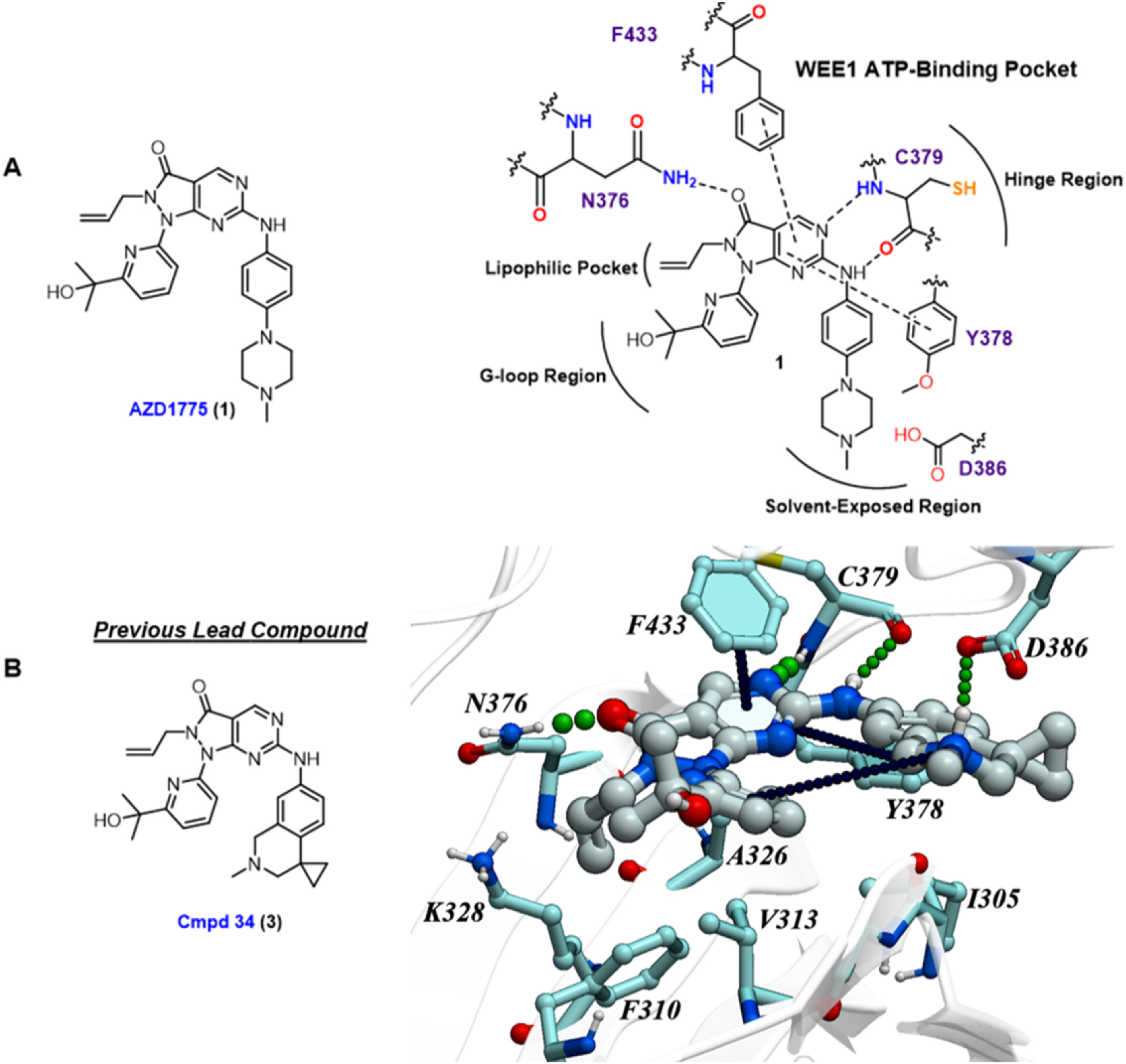
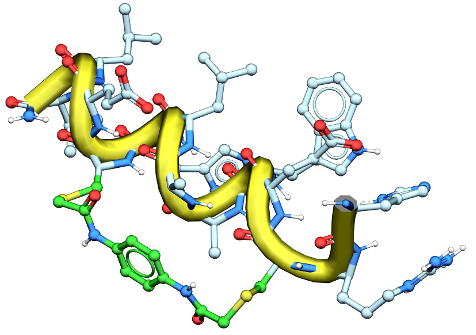
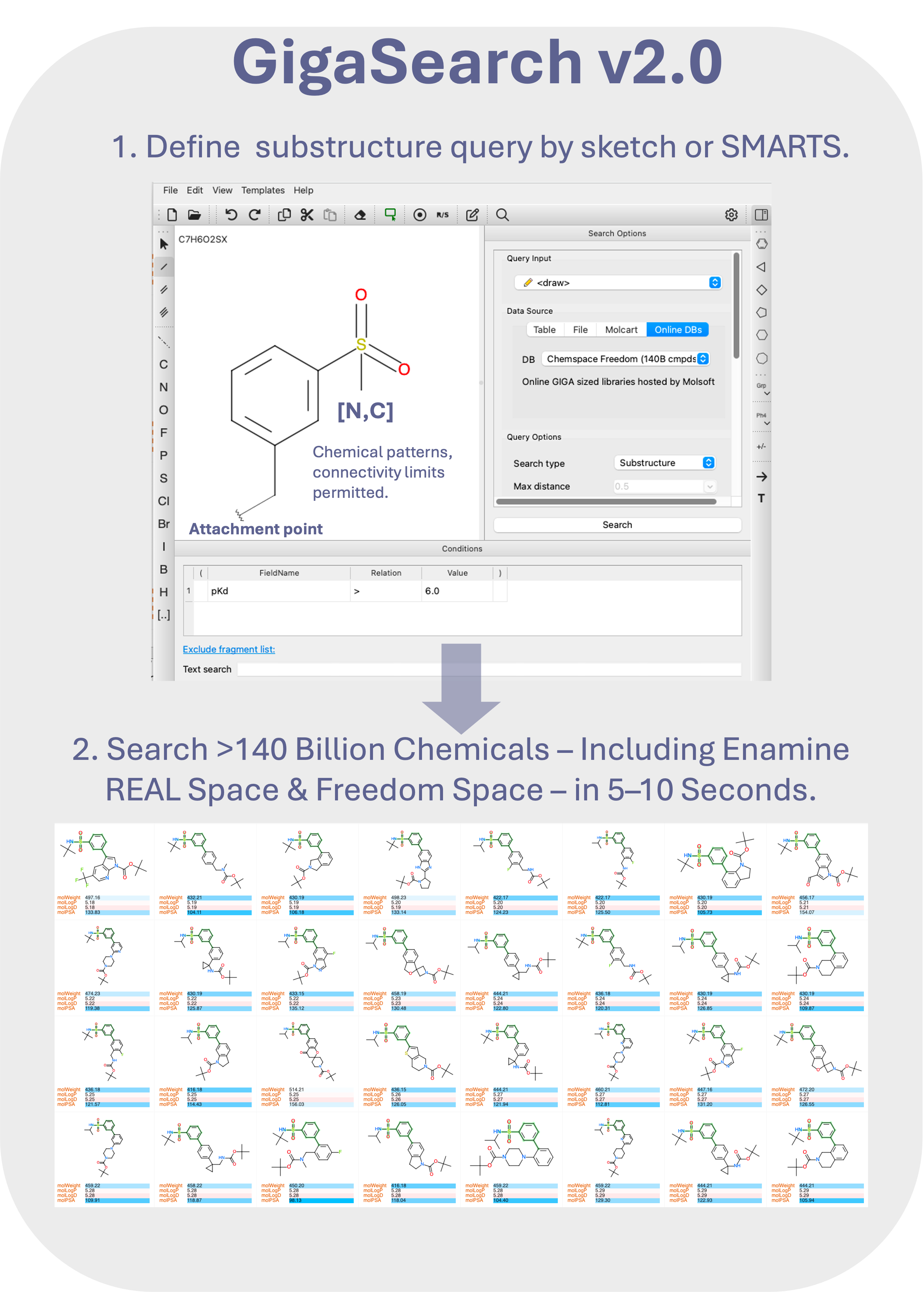
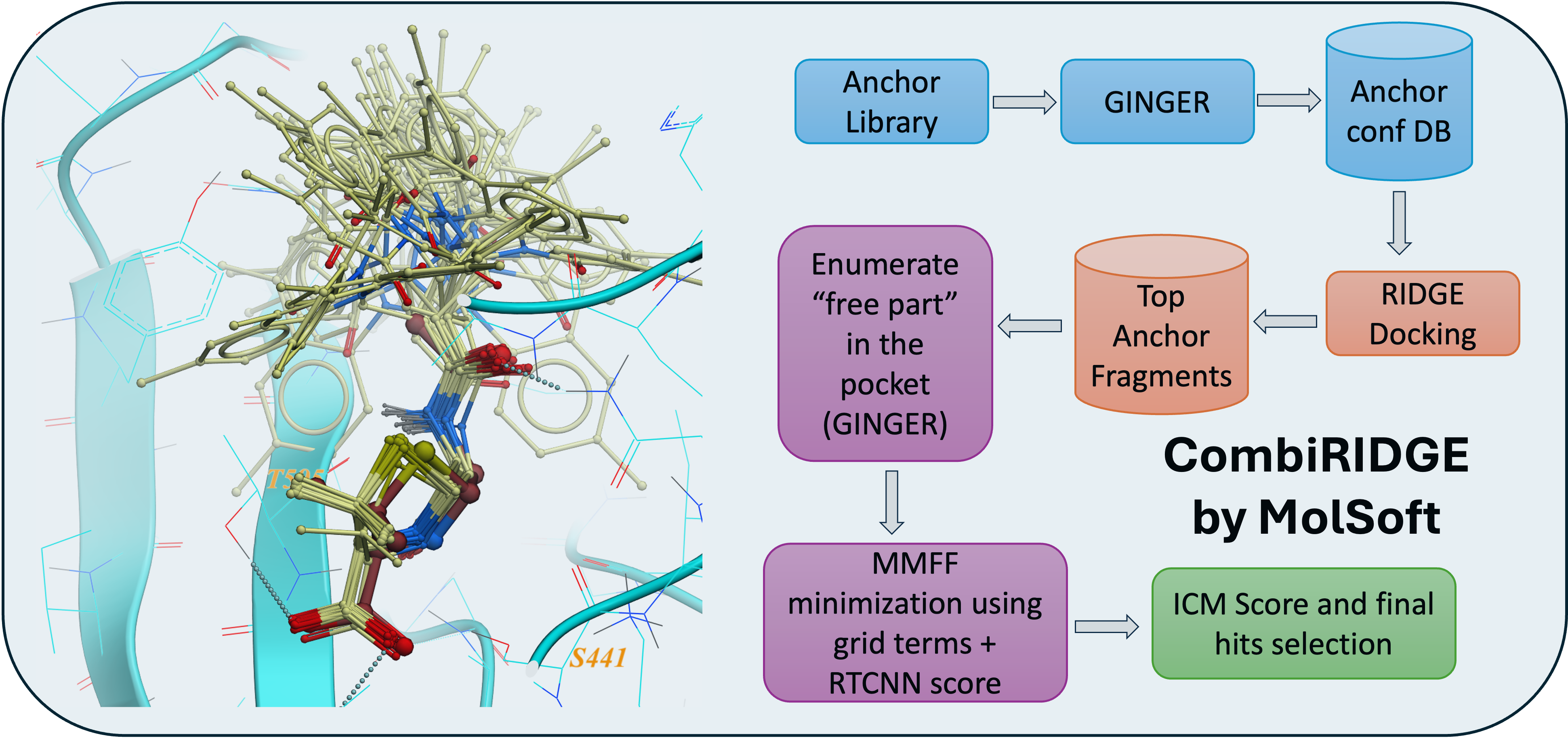
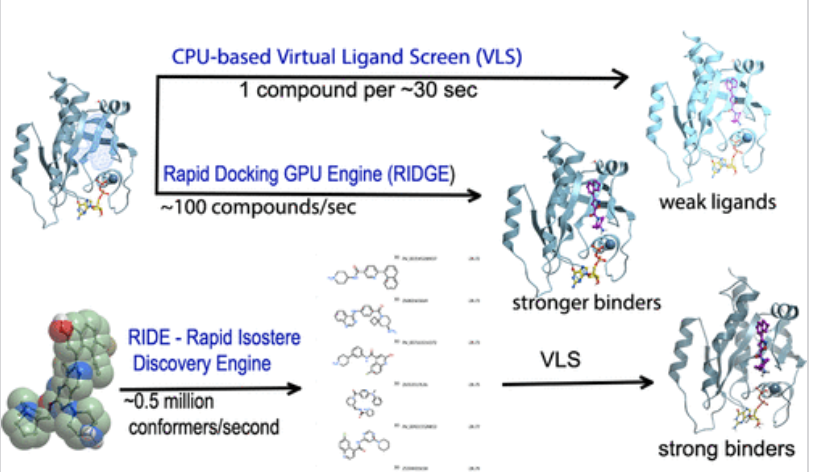
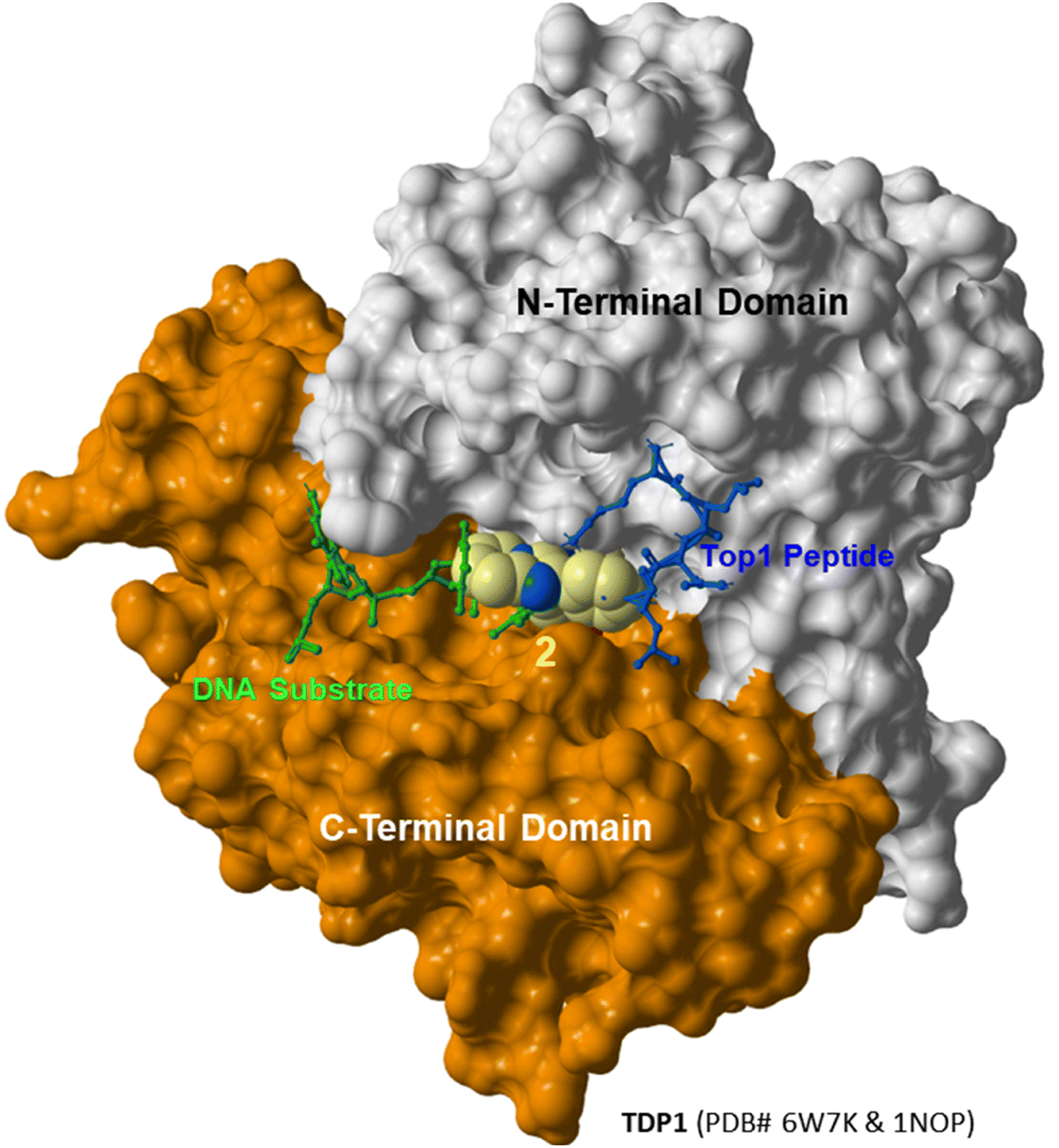

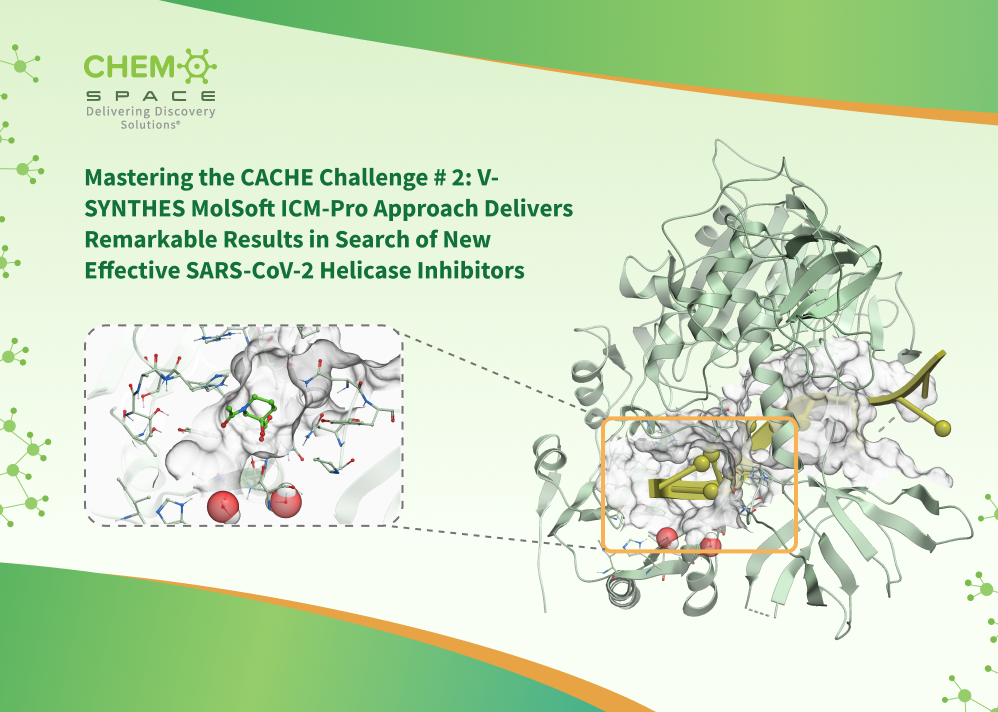
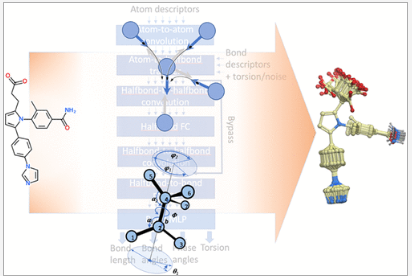

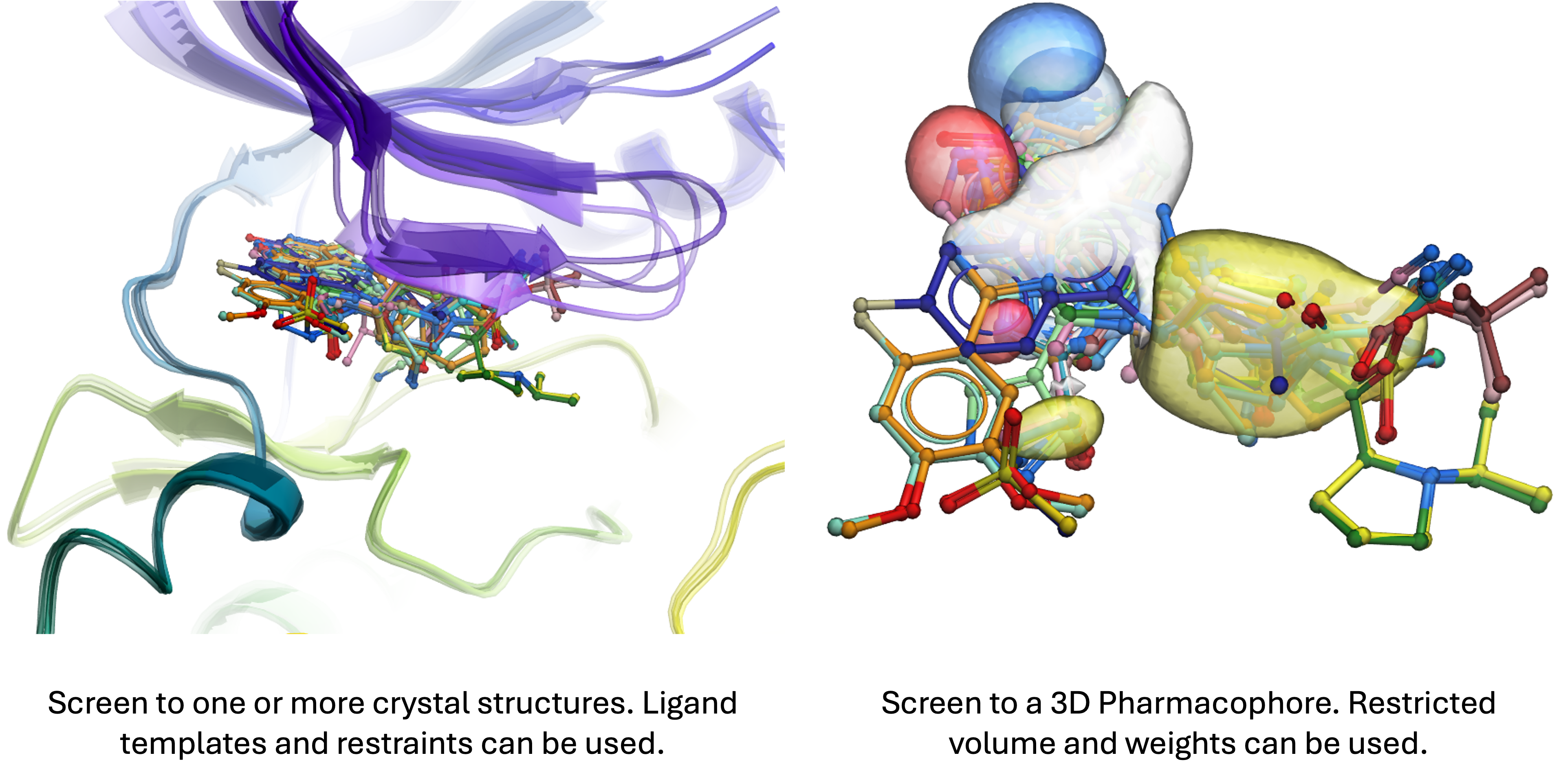

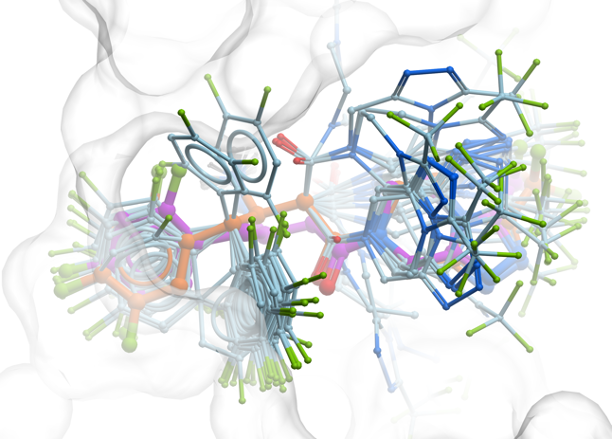



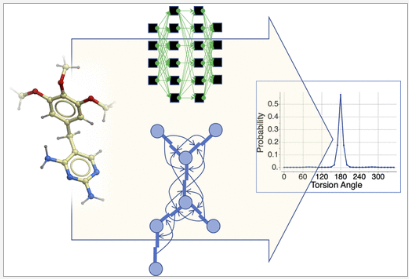


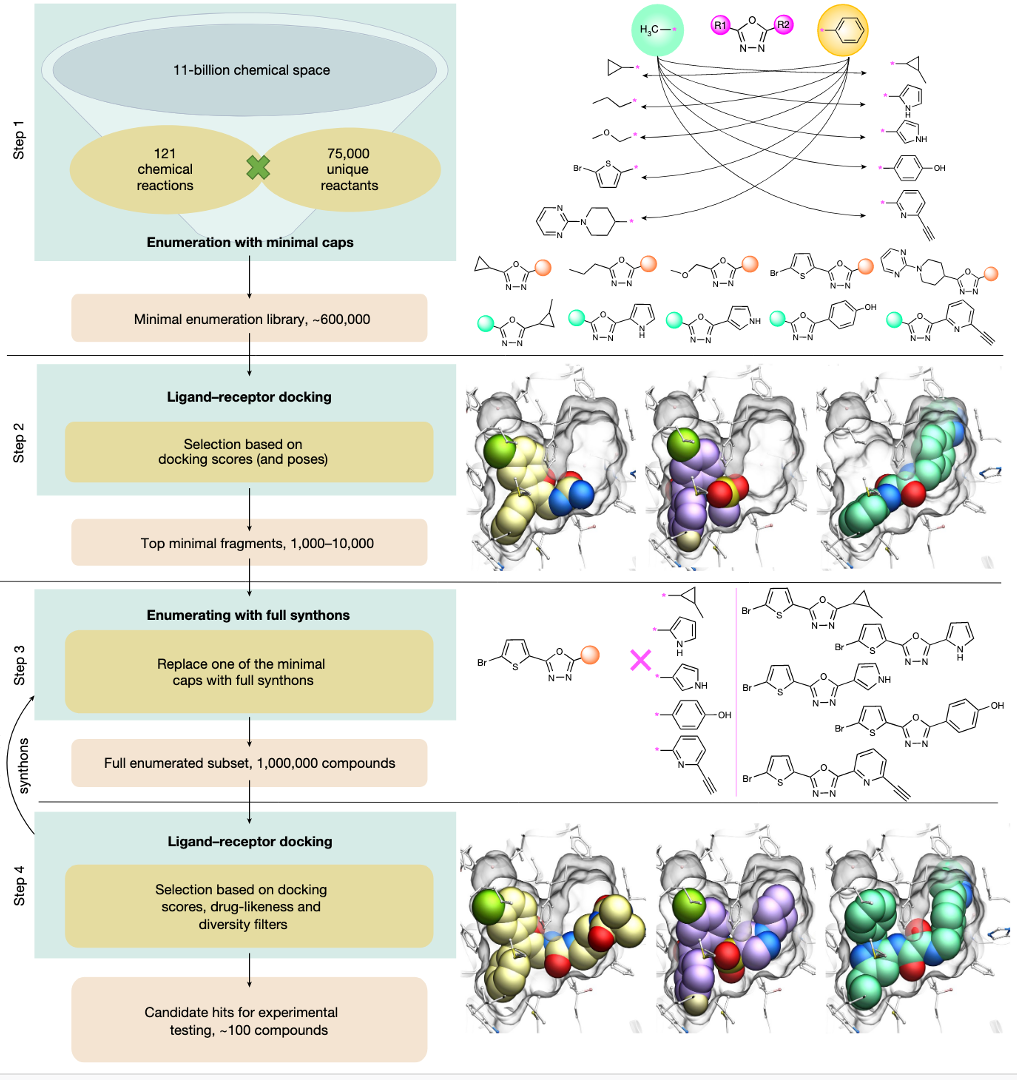


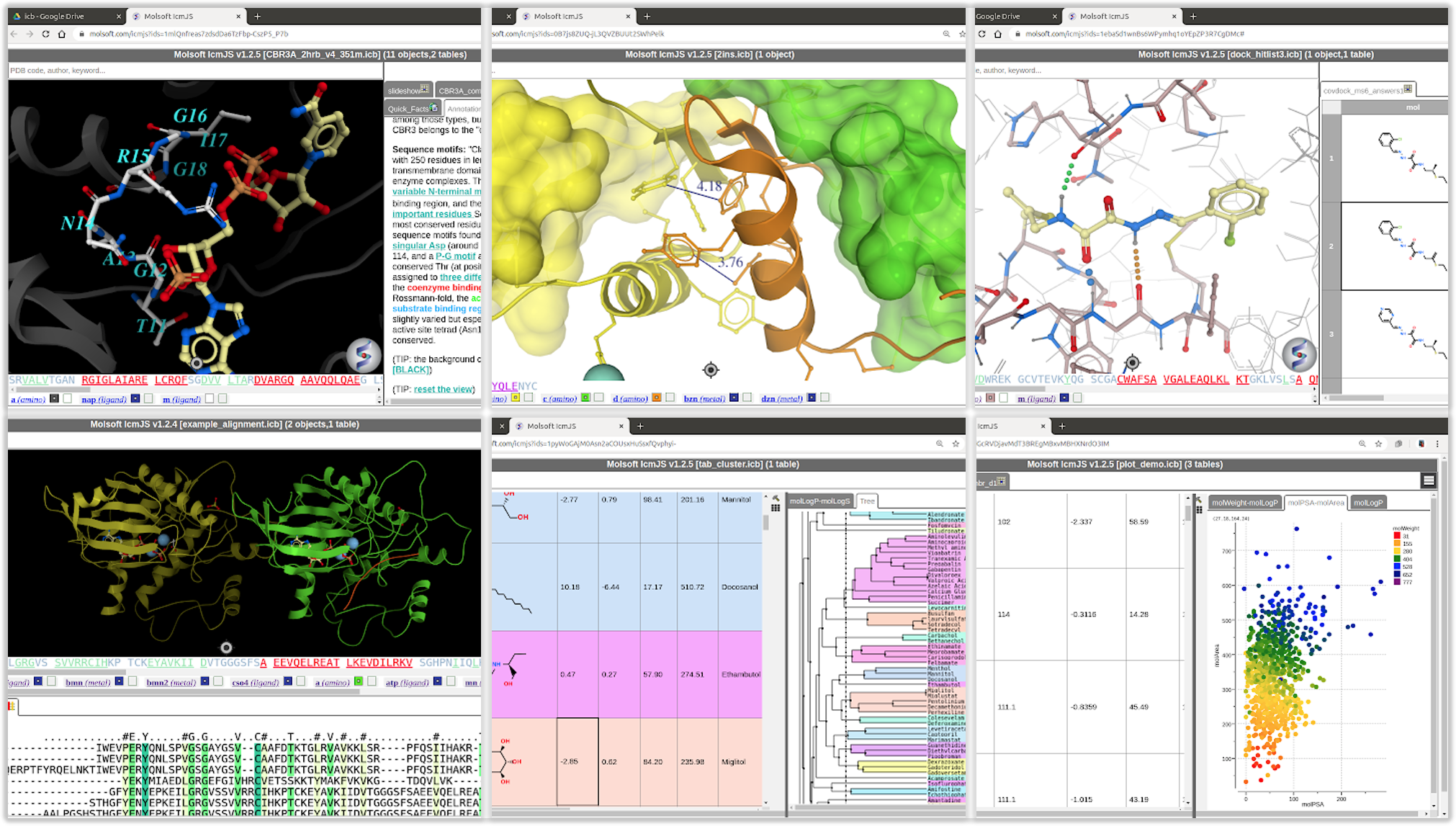
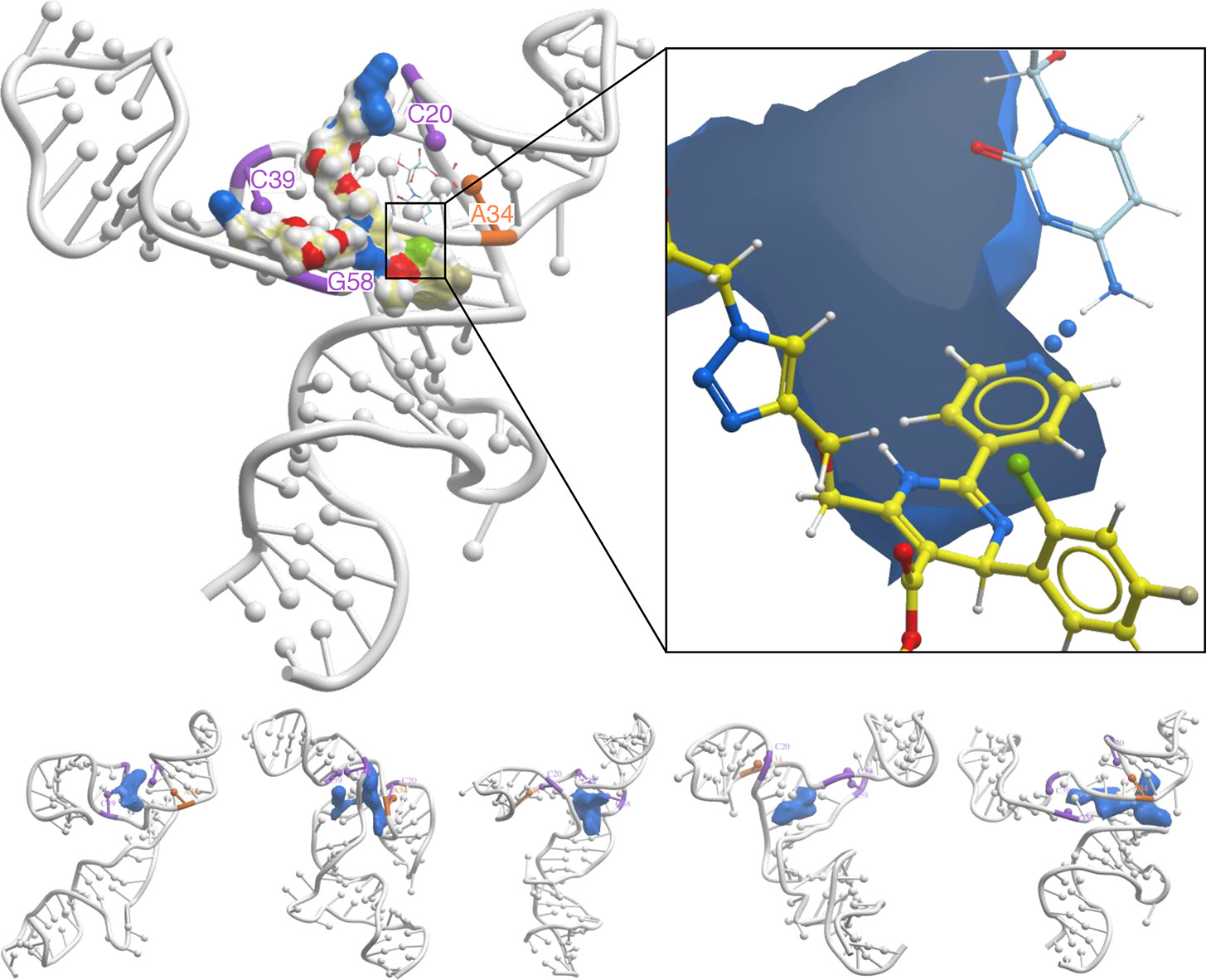
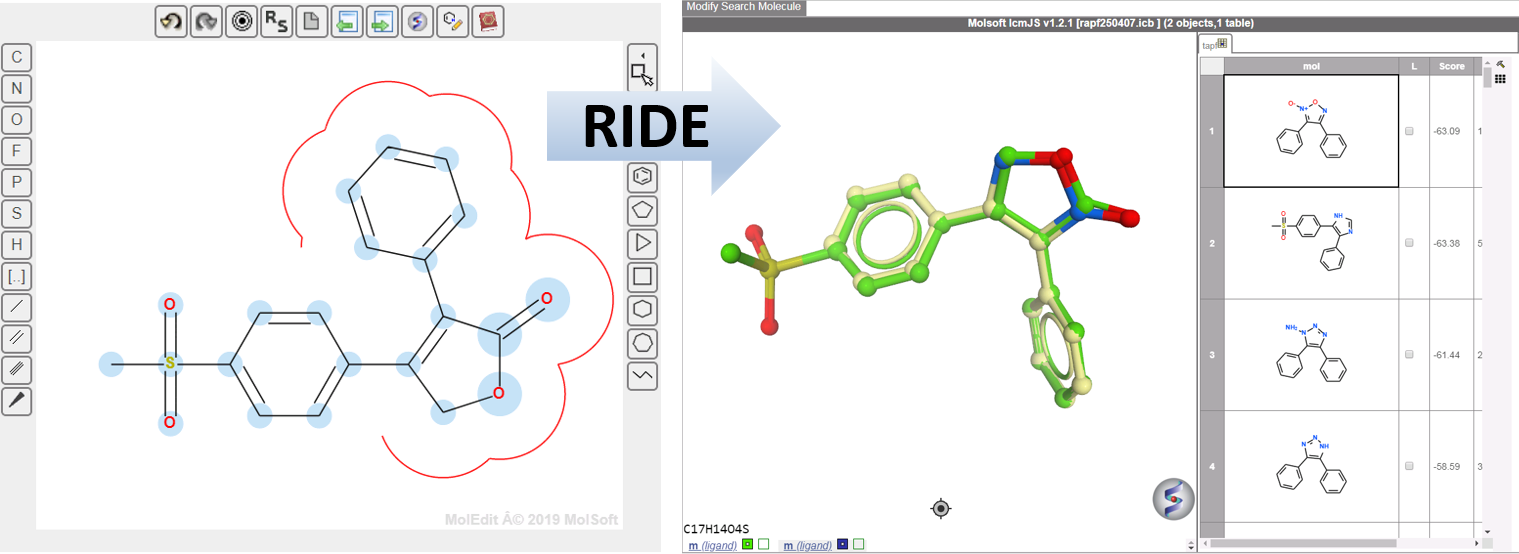
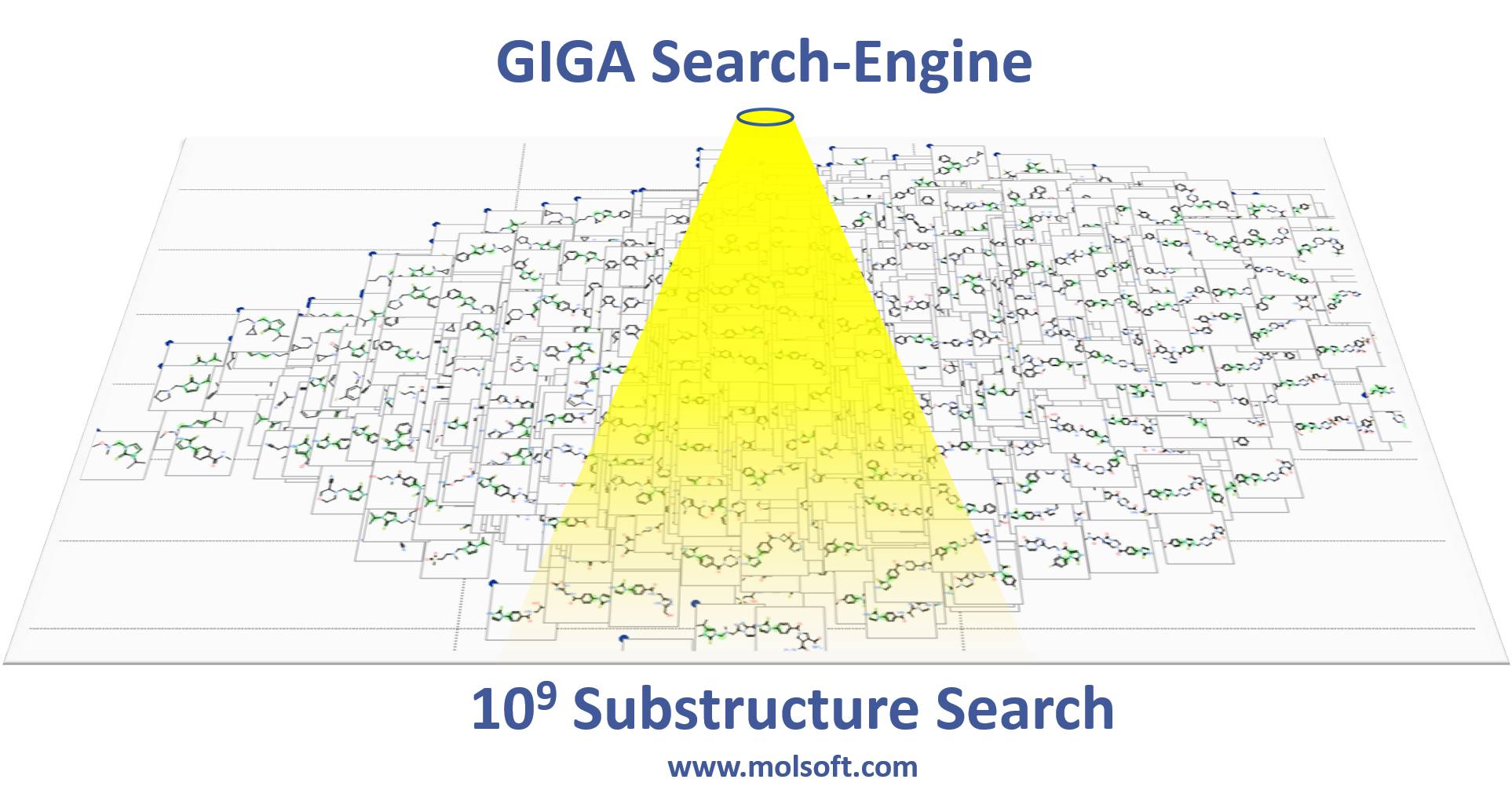
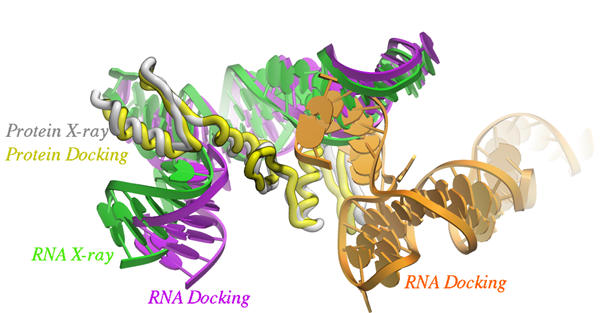
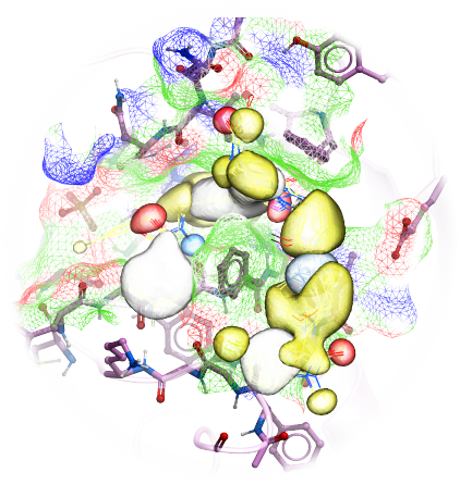
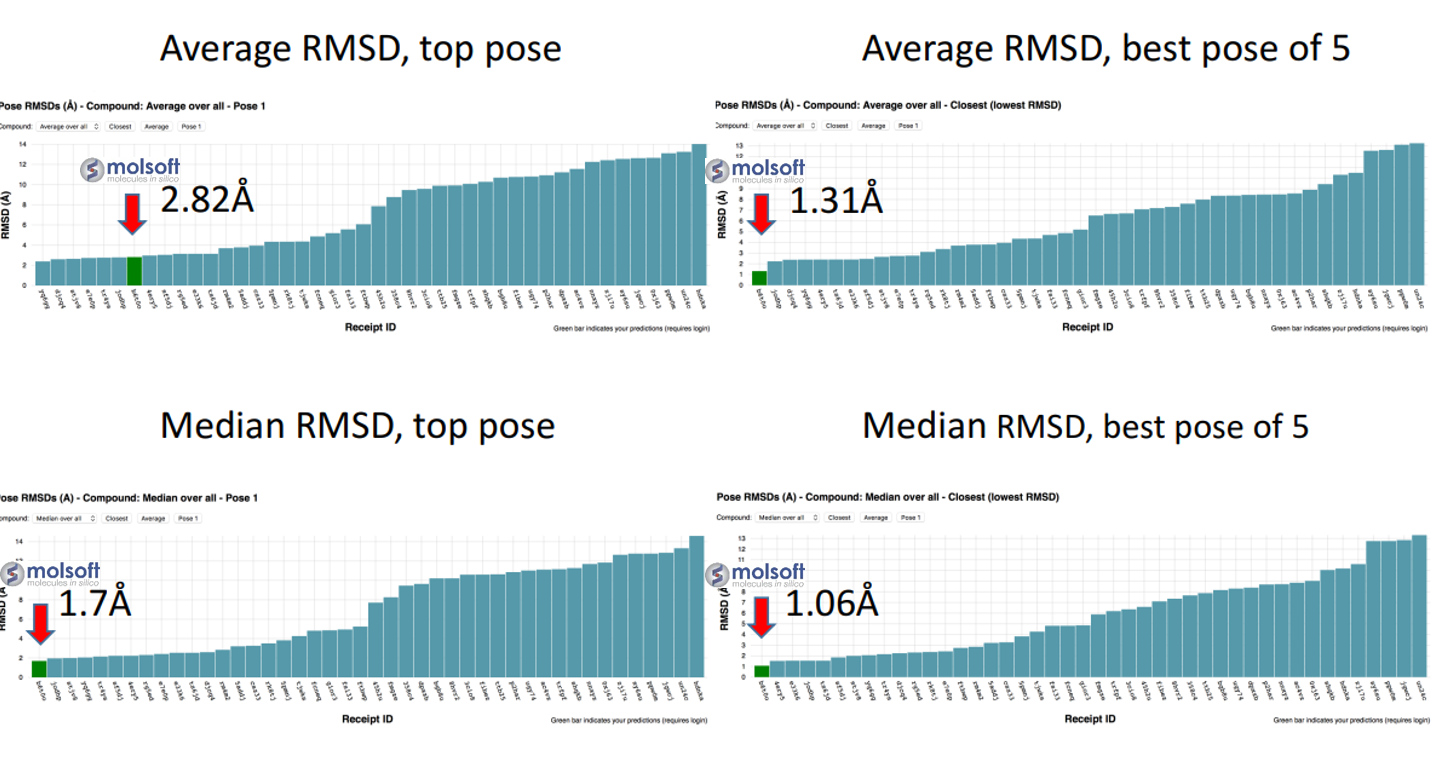
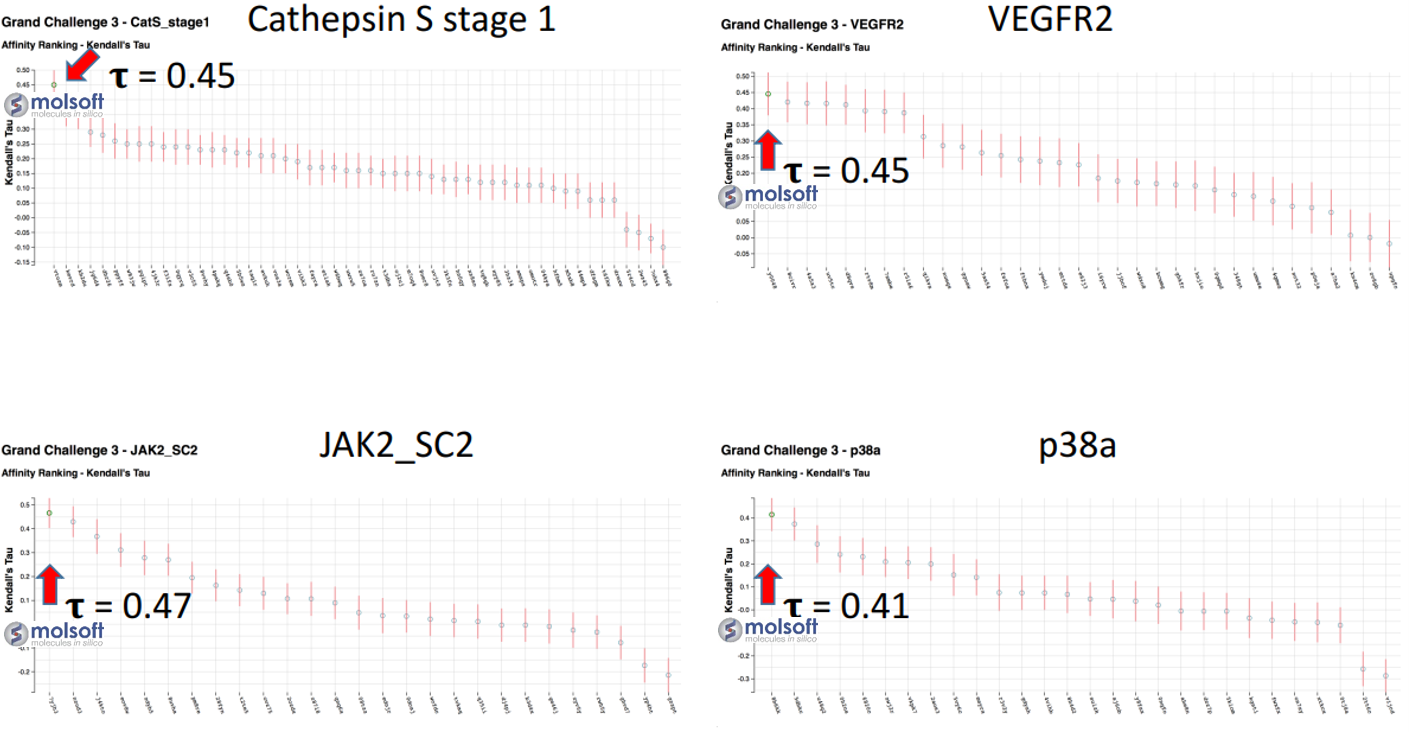
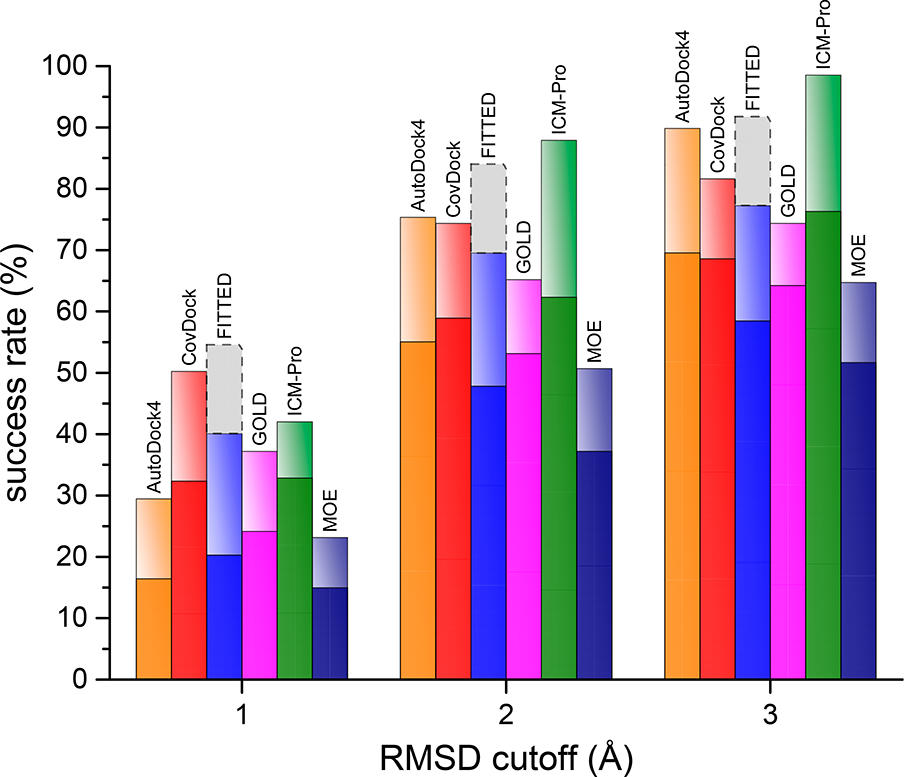

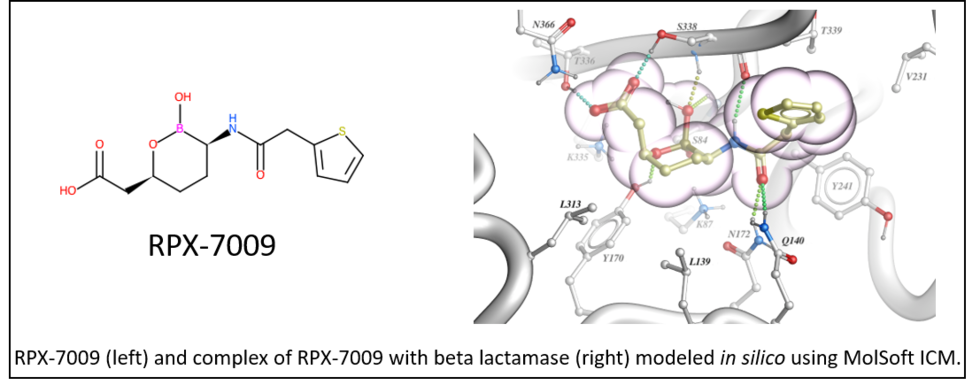
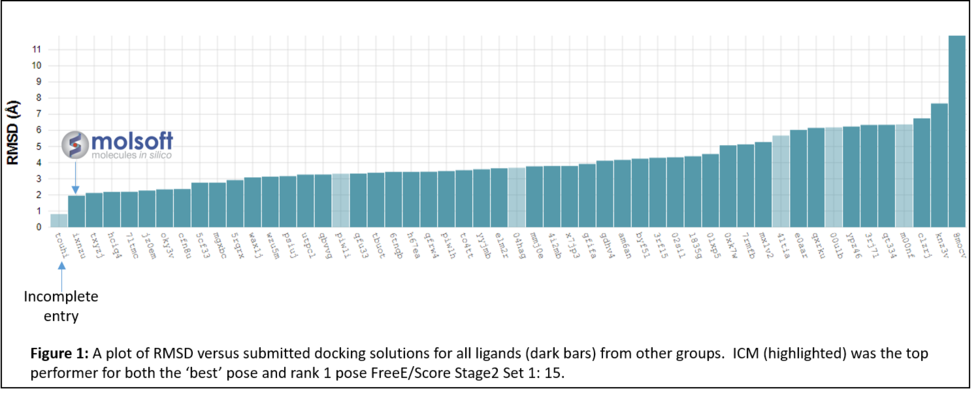
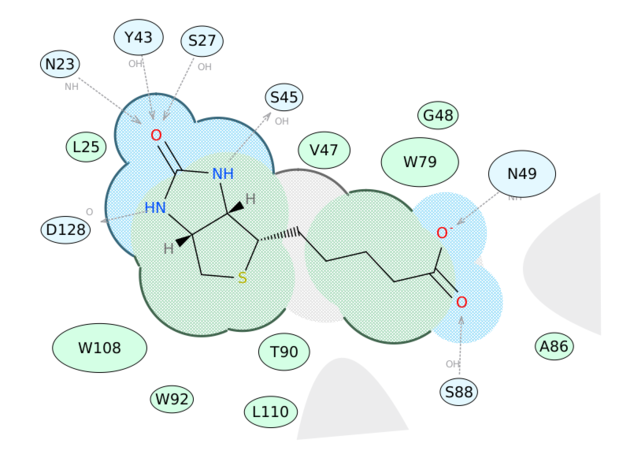
 A new paper in
A new paper in 

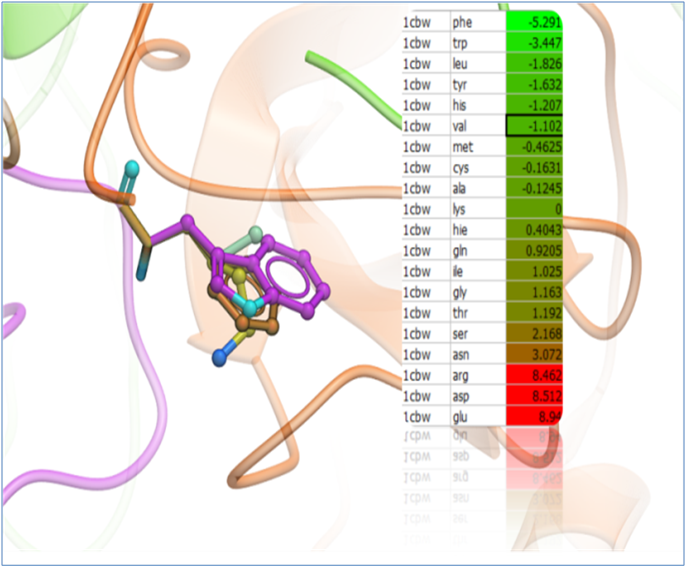
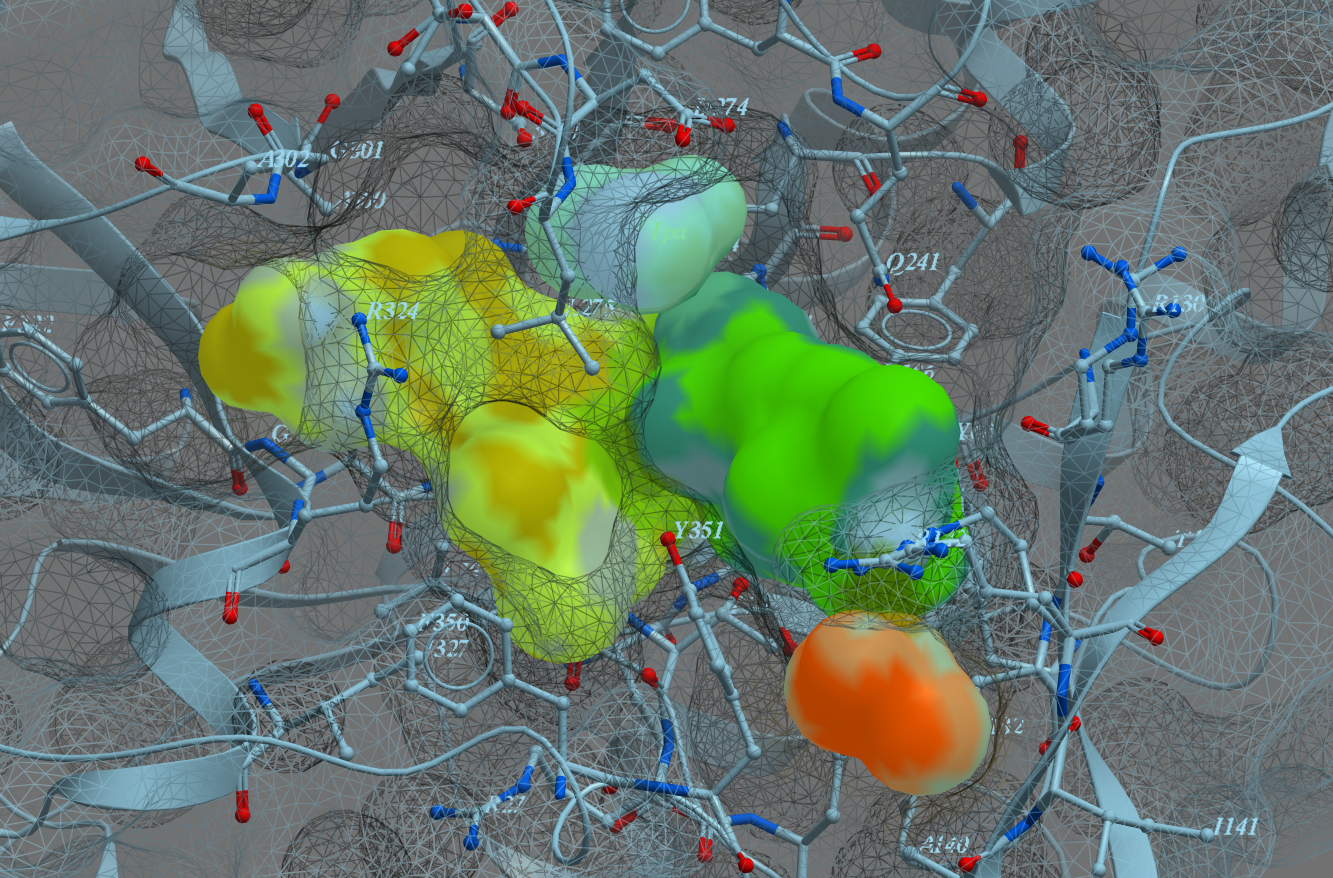
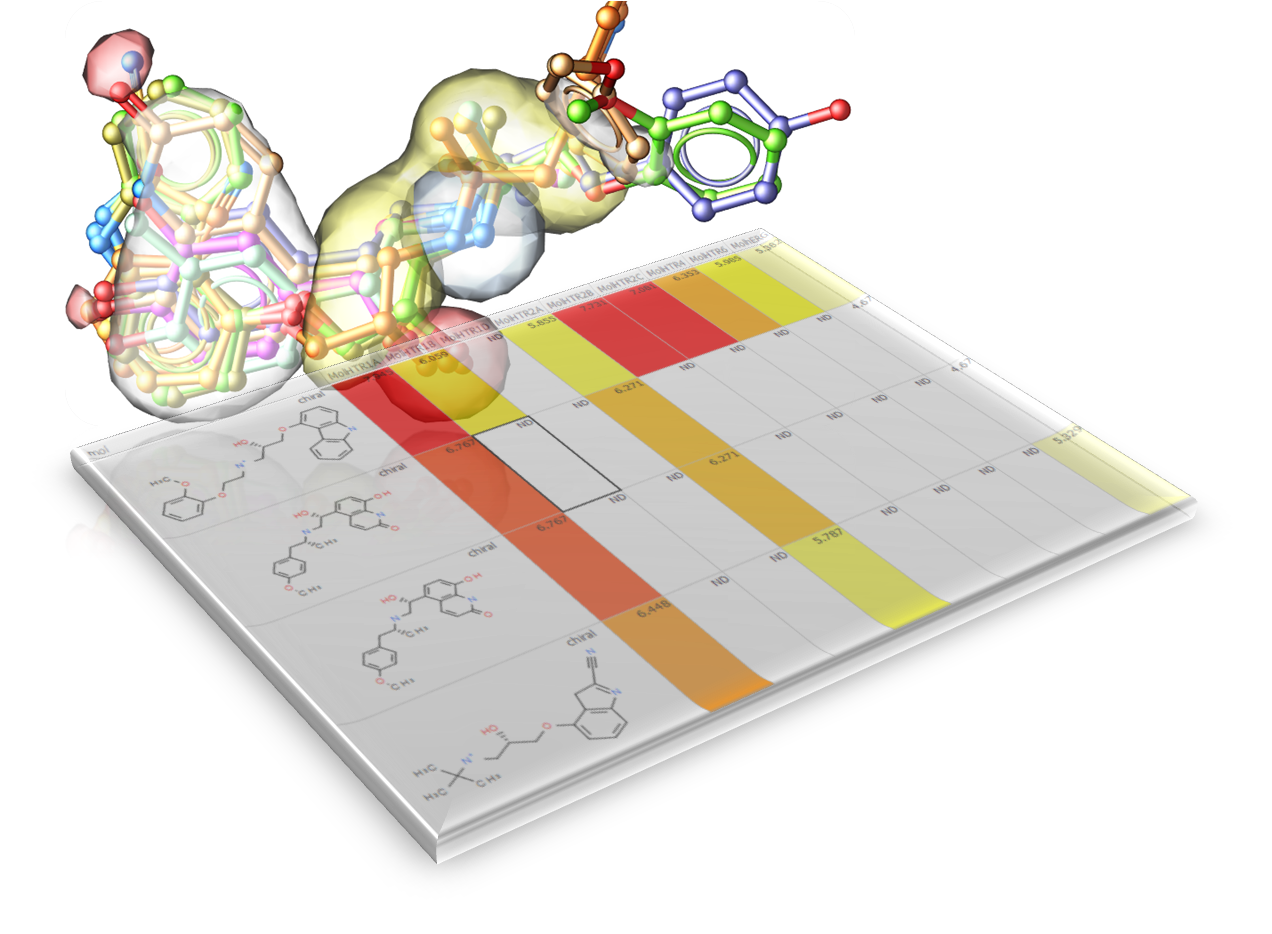
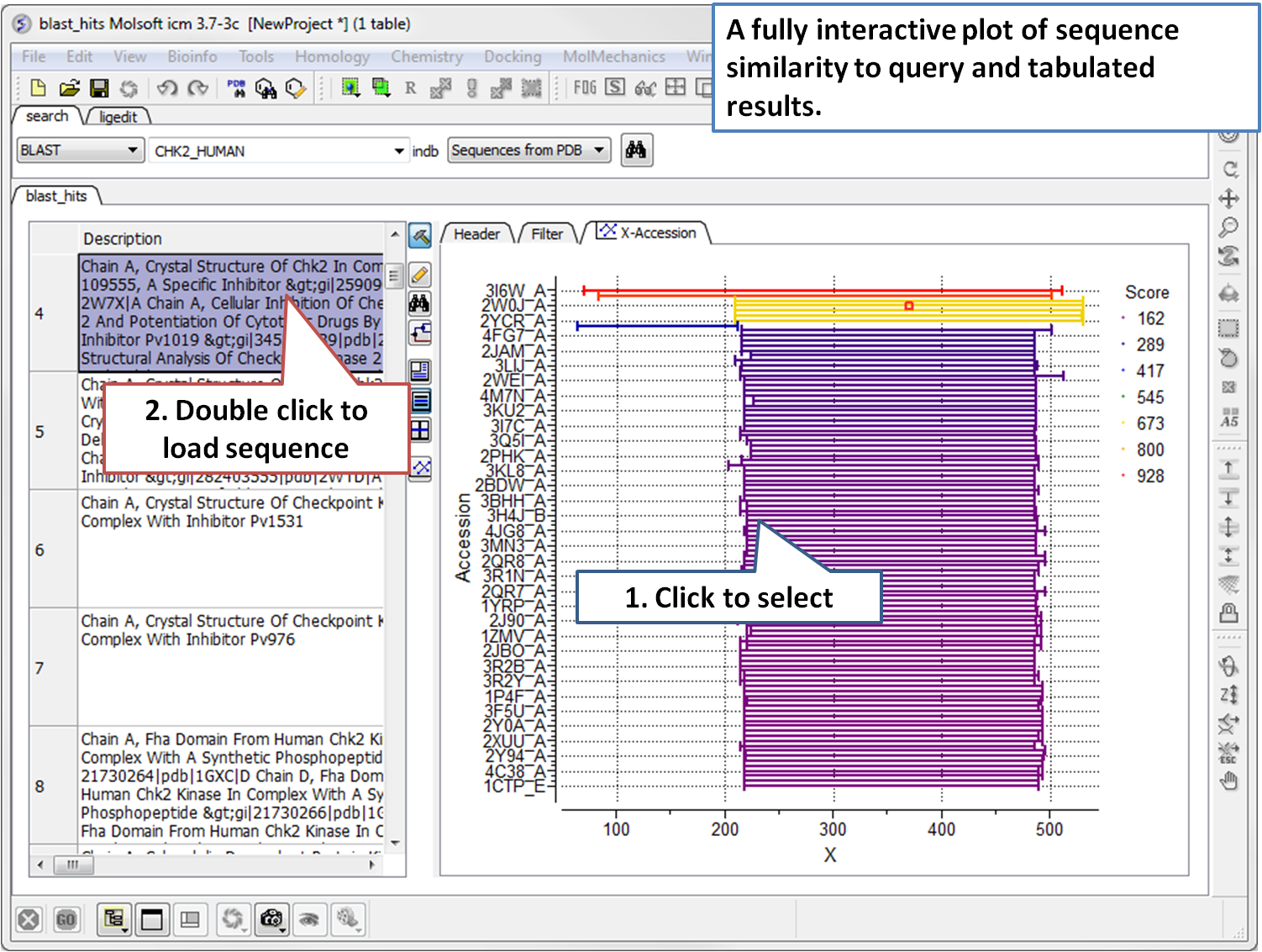
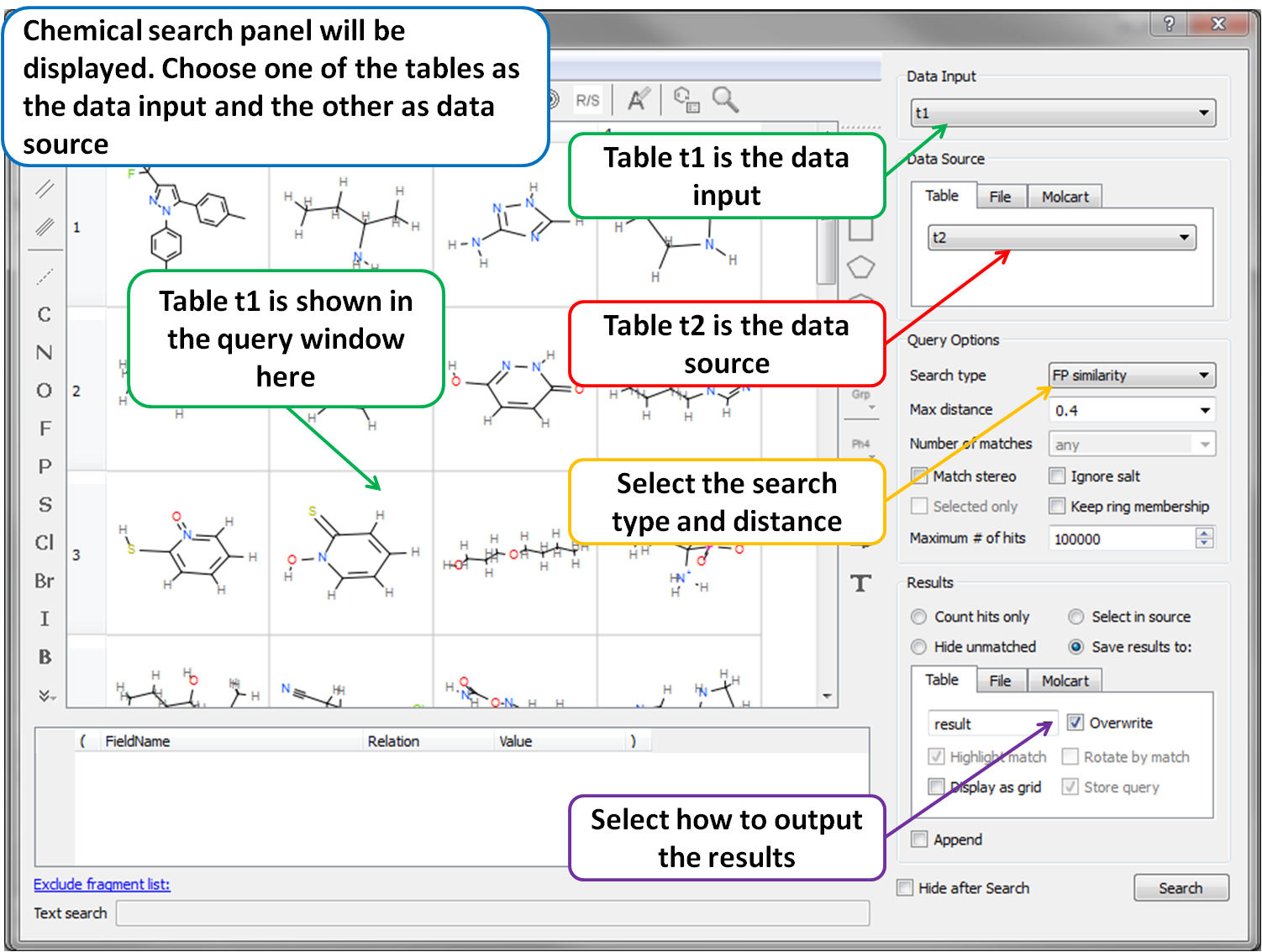


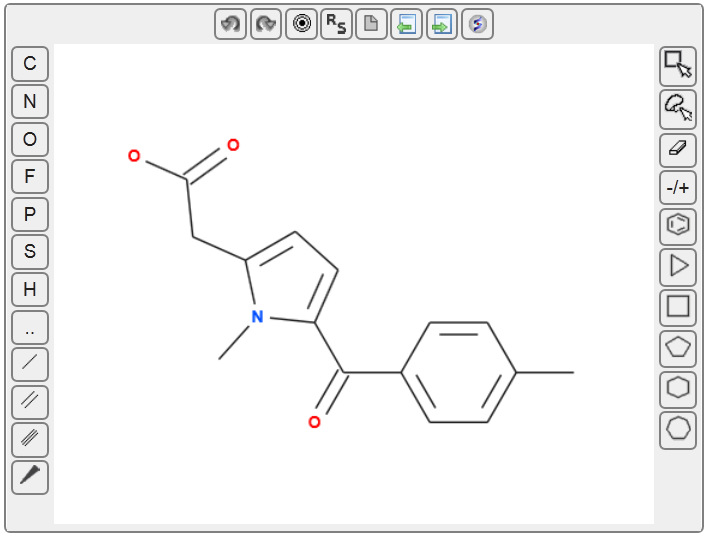

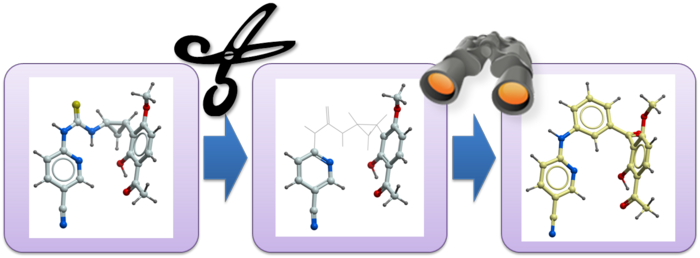
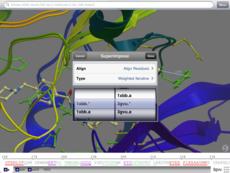

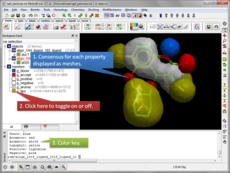
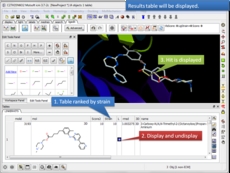
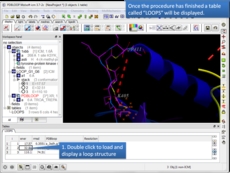




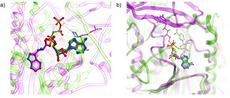


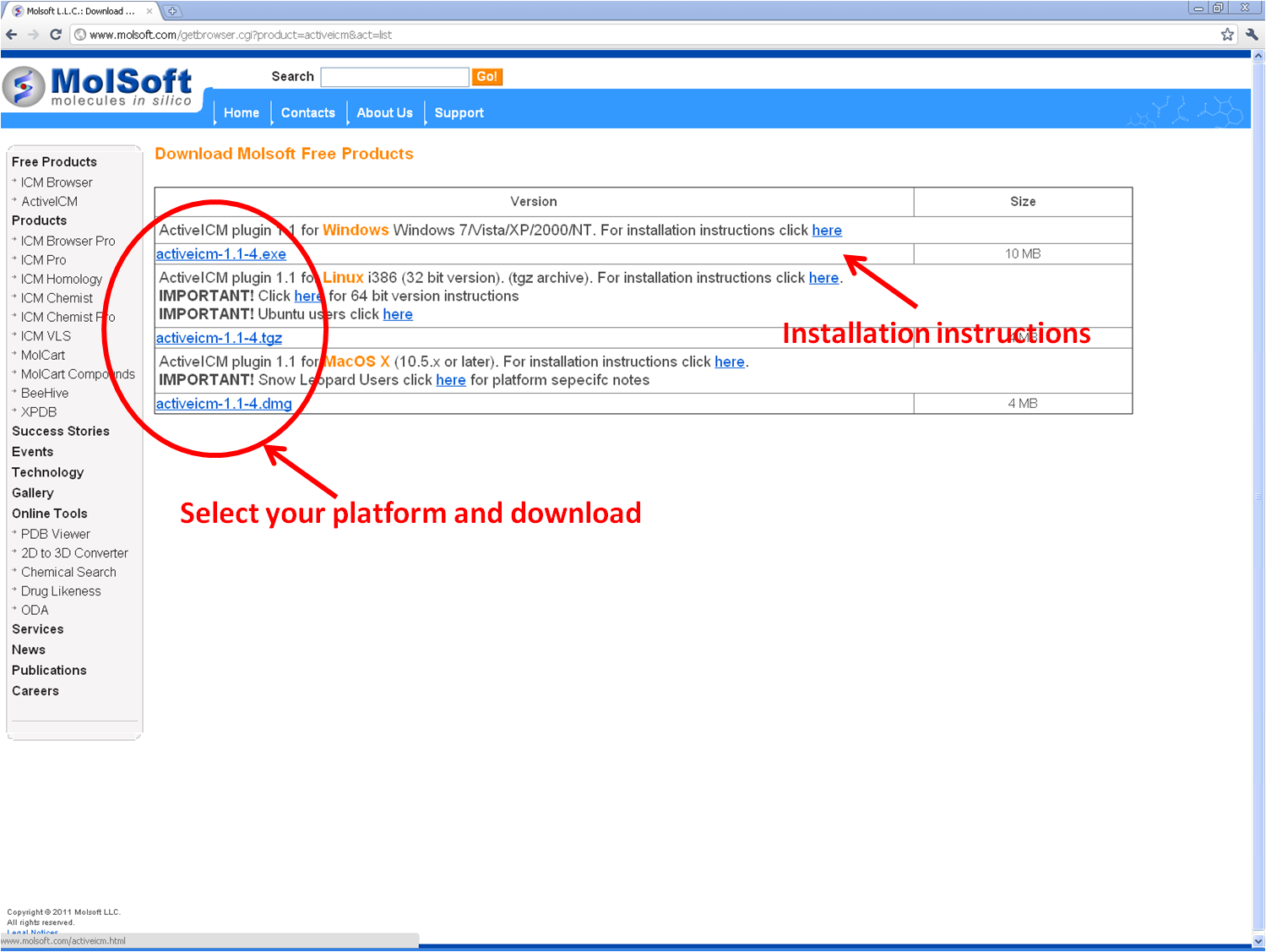

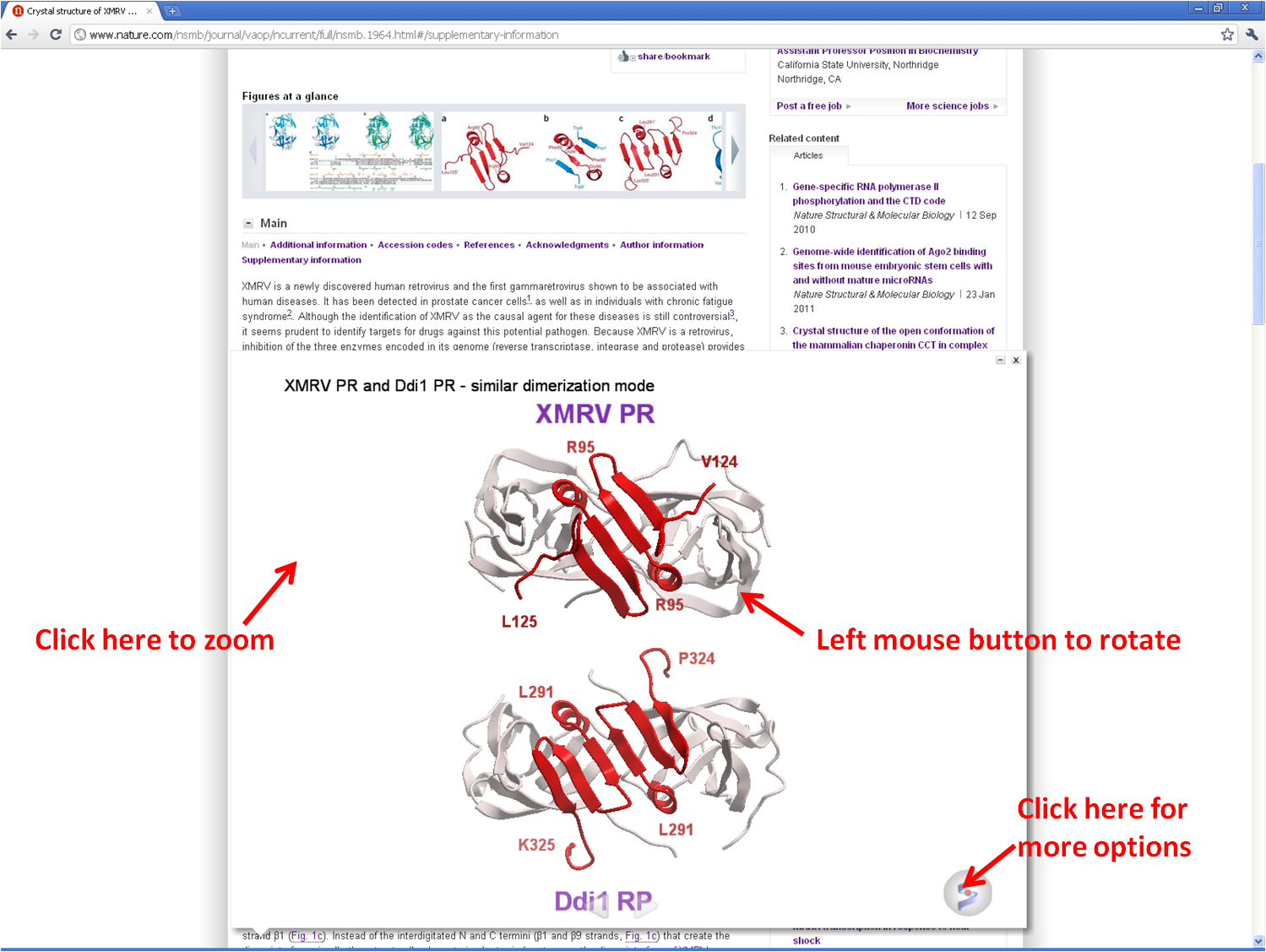



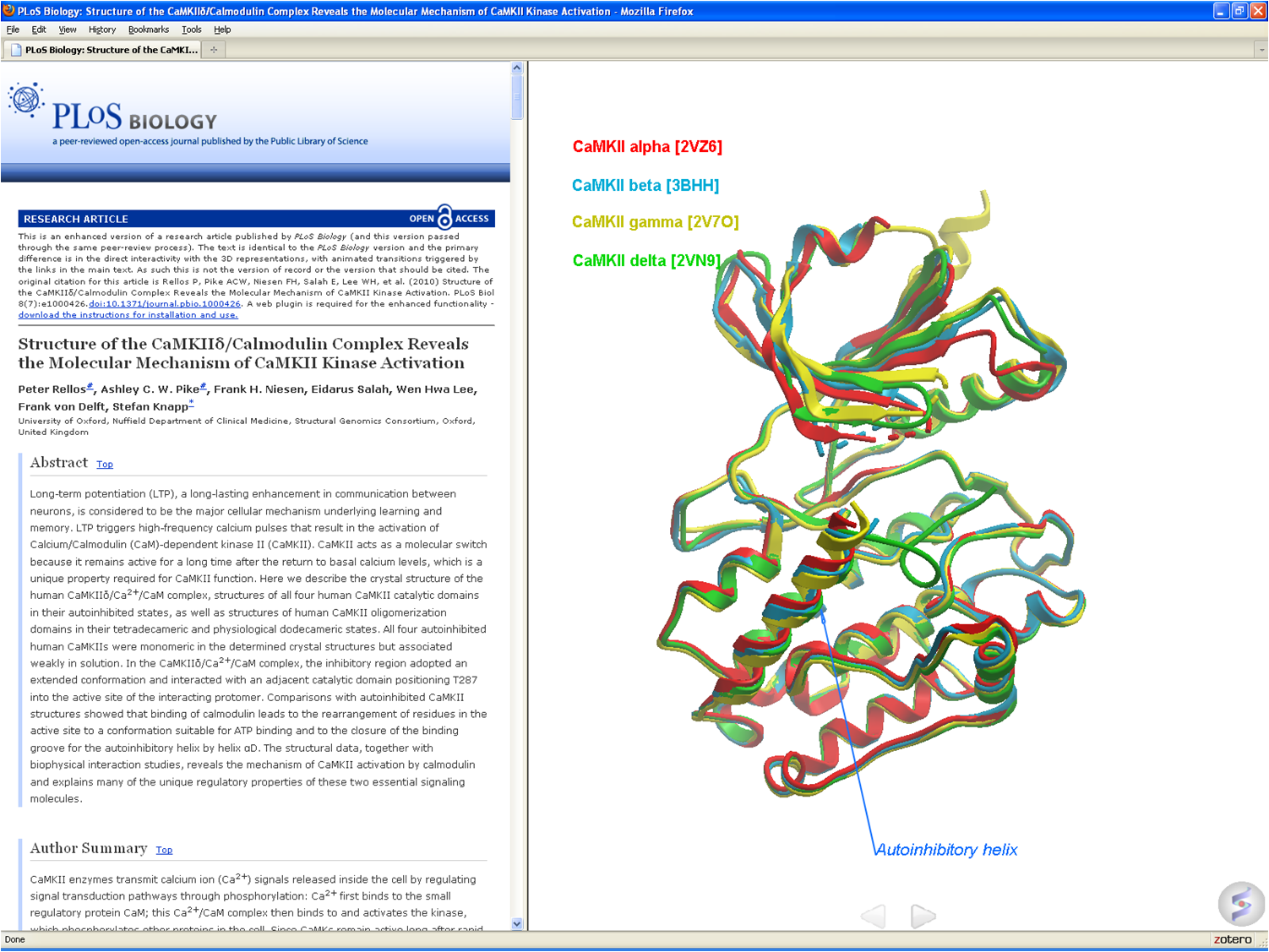

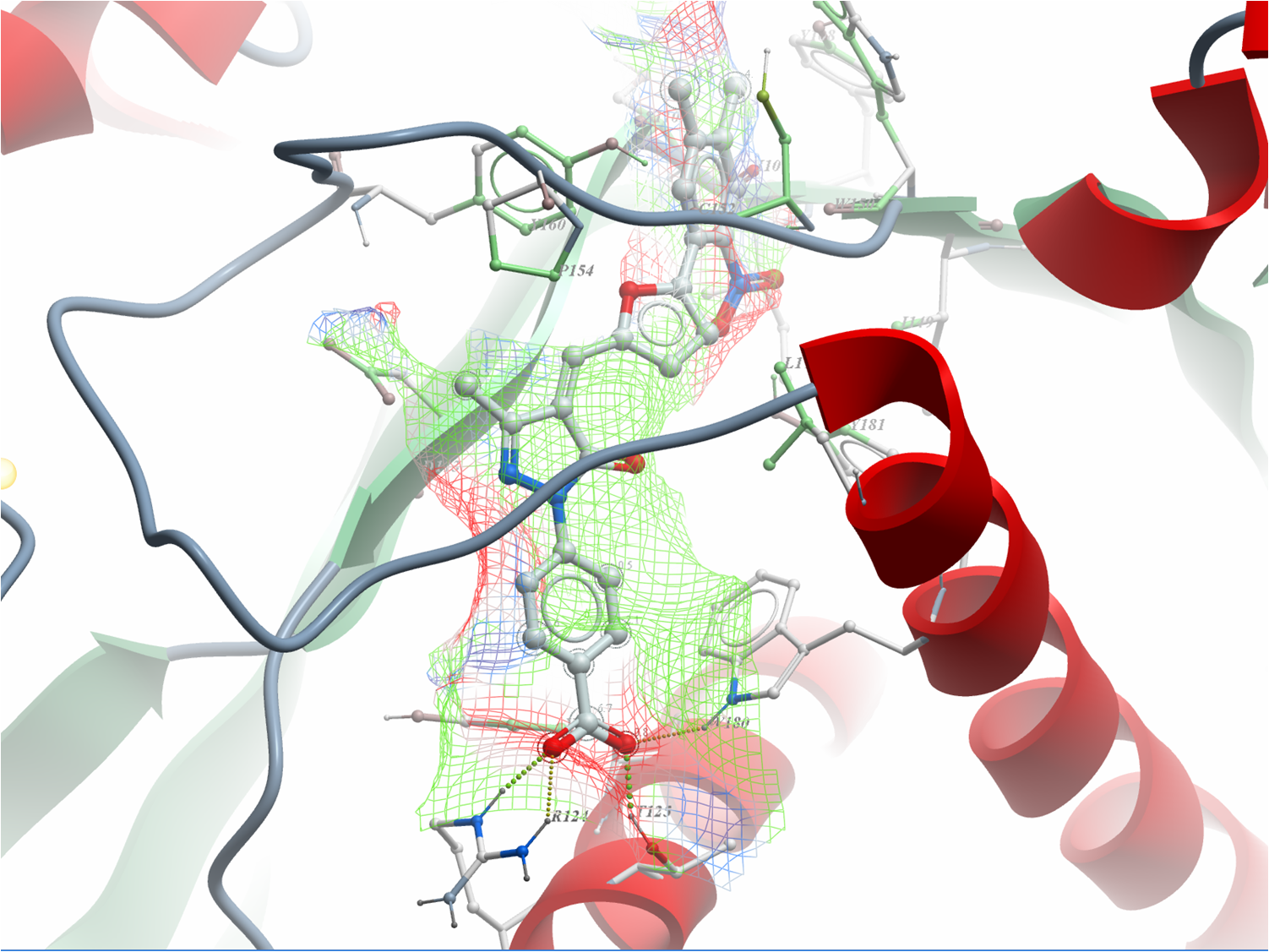


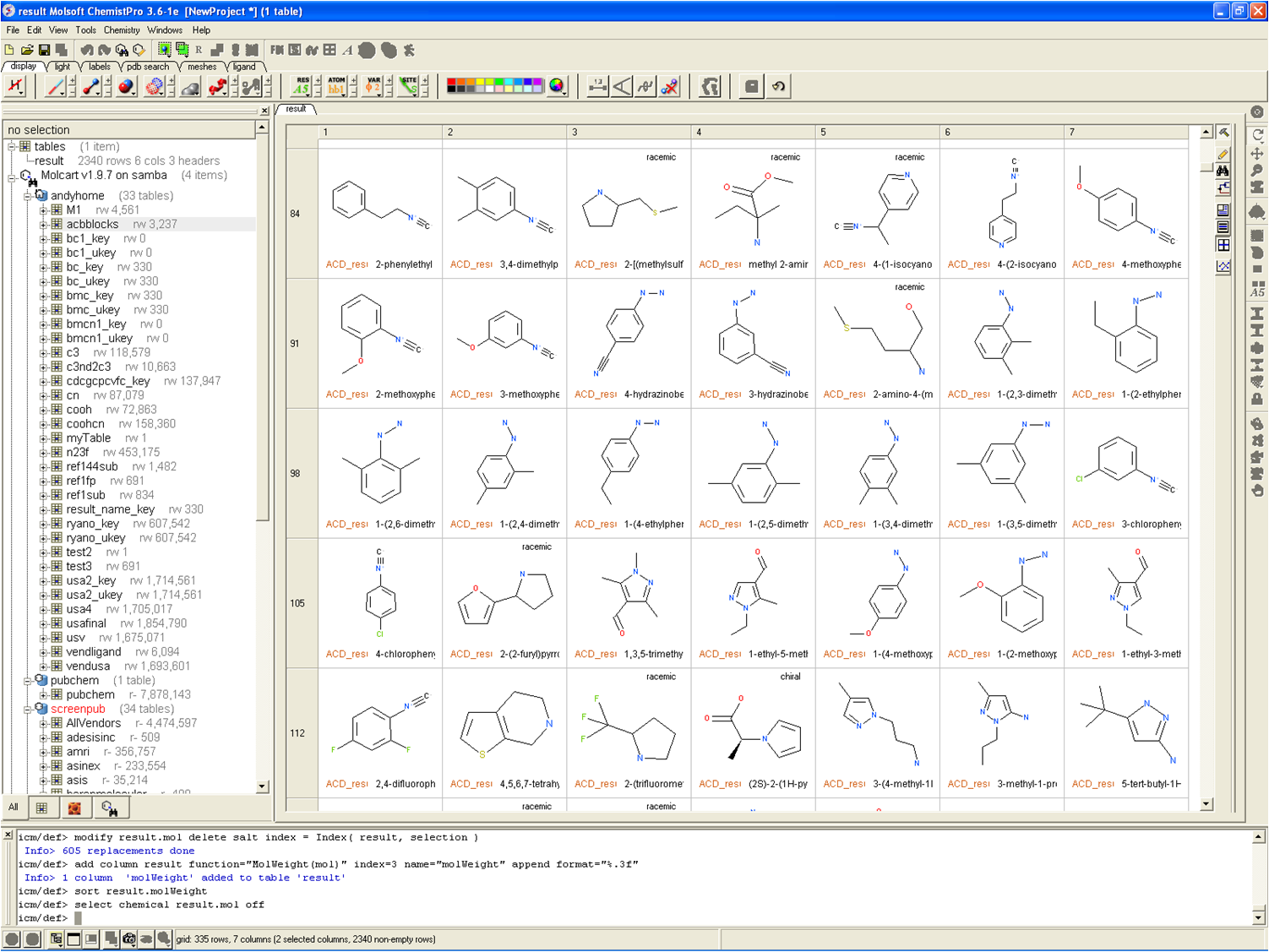

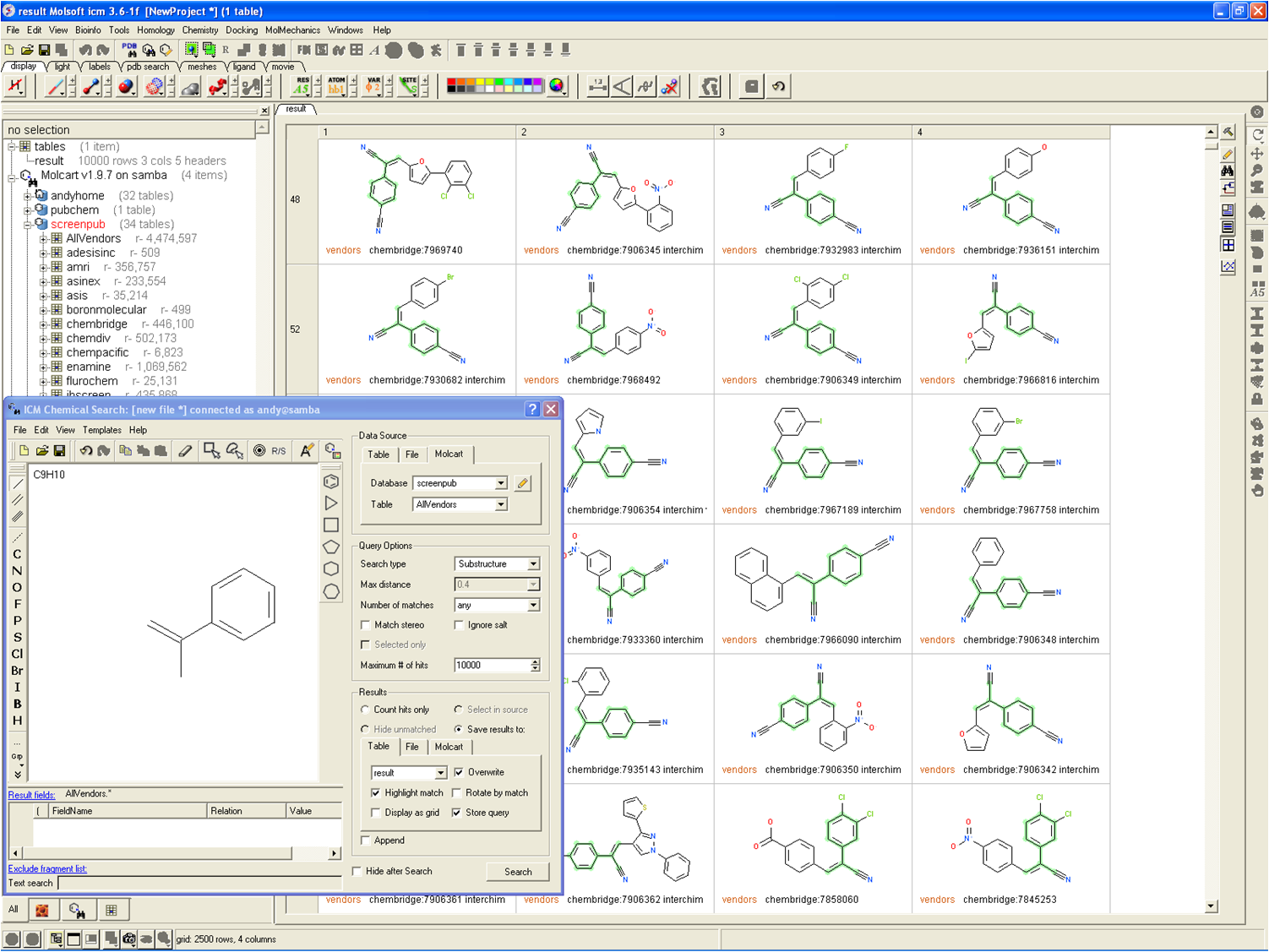
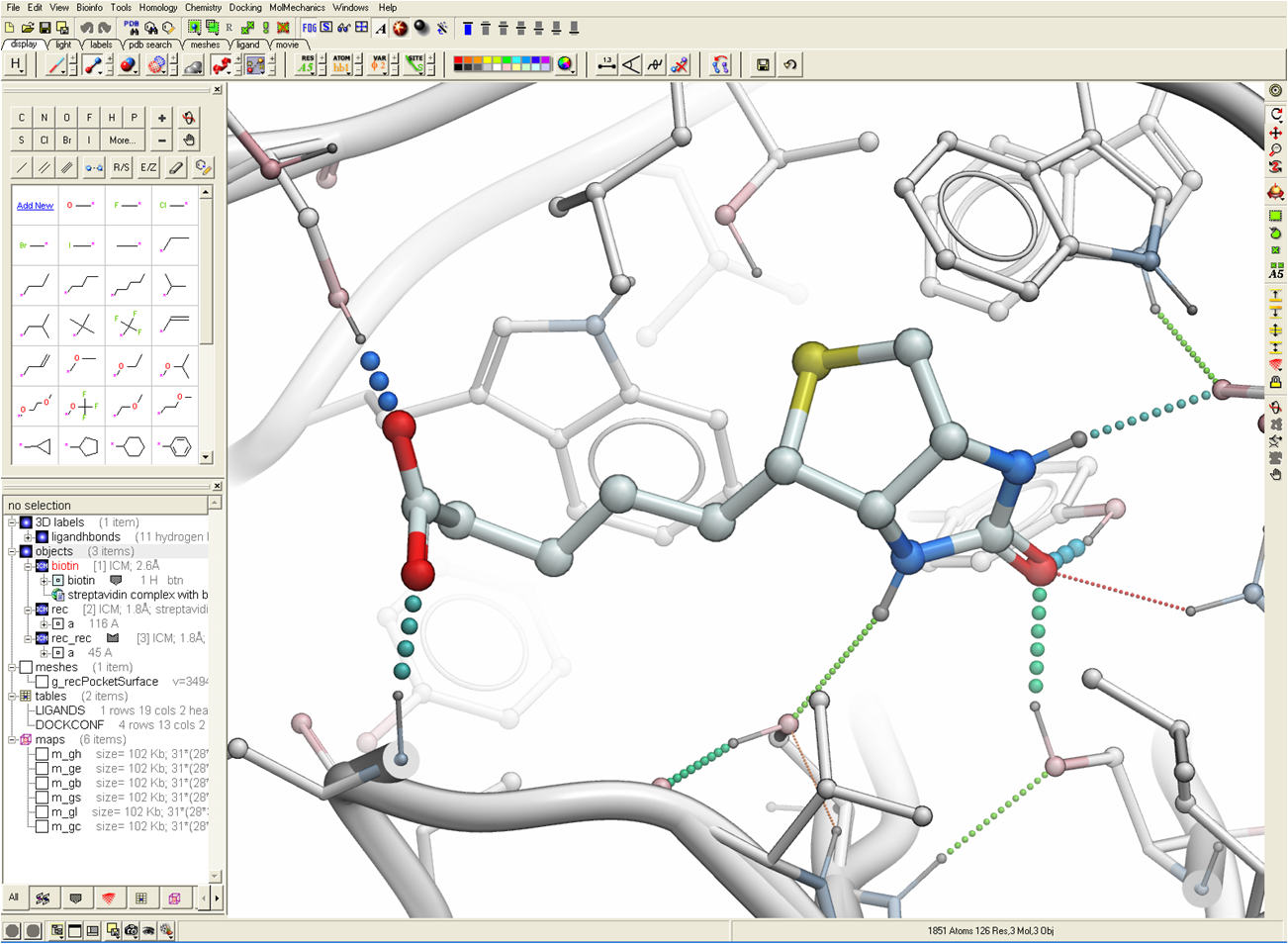
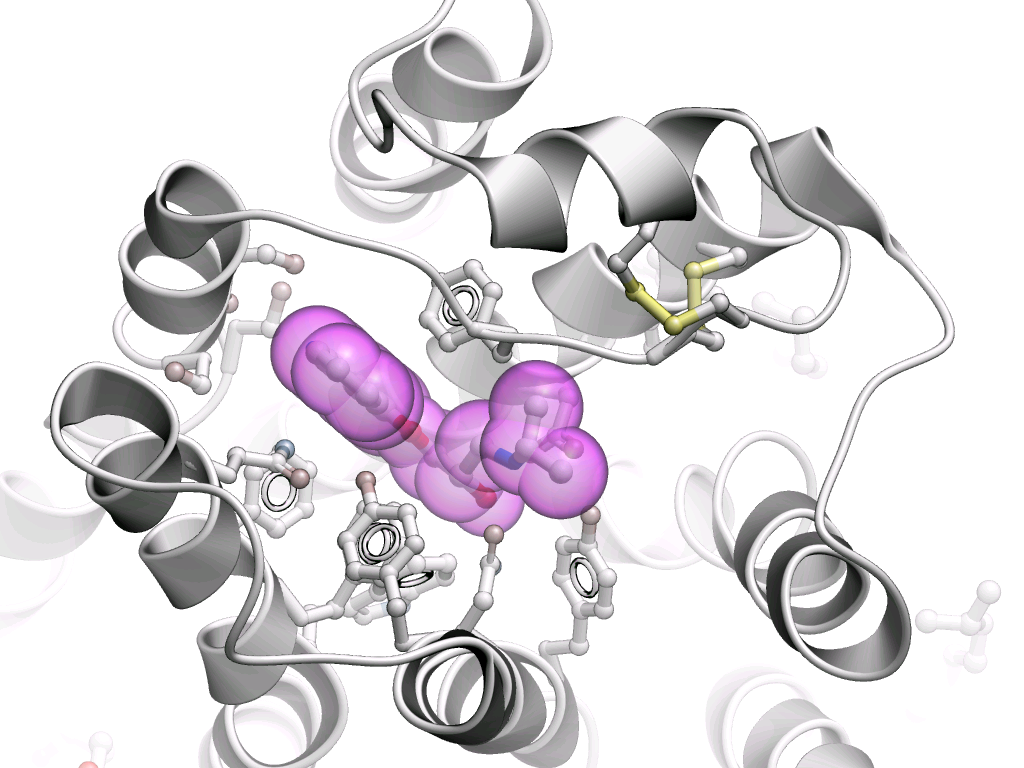







 We now have over 80 members in the ICM Discussion Forum but this only represents a very small percentage of the ICM users worldwide. Click
We now have over 80 members in the ICM Discussion Forum but this only represents a very small percentage of the ICM users worldwide. Click 
 You can now read, render and display any 3D object in ICM. e.g. KMZ and COLLADA format directly from the google sketchup website. We have also added options for changing the texture and material of a 3D object. See
You can now read, render and display any 3D object in ICM. e.g. KMZ and COLLADA format directly from the google sketchup website. We have also added options for changing the texture and material of a 3D object. See 
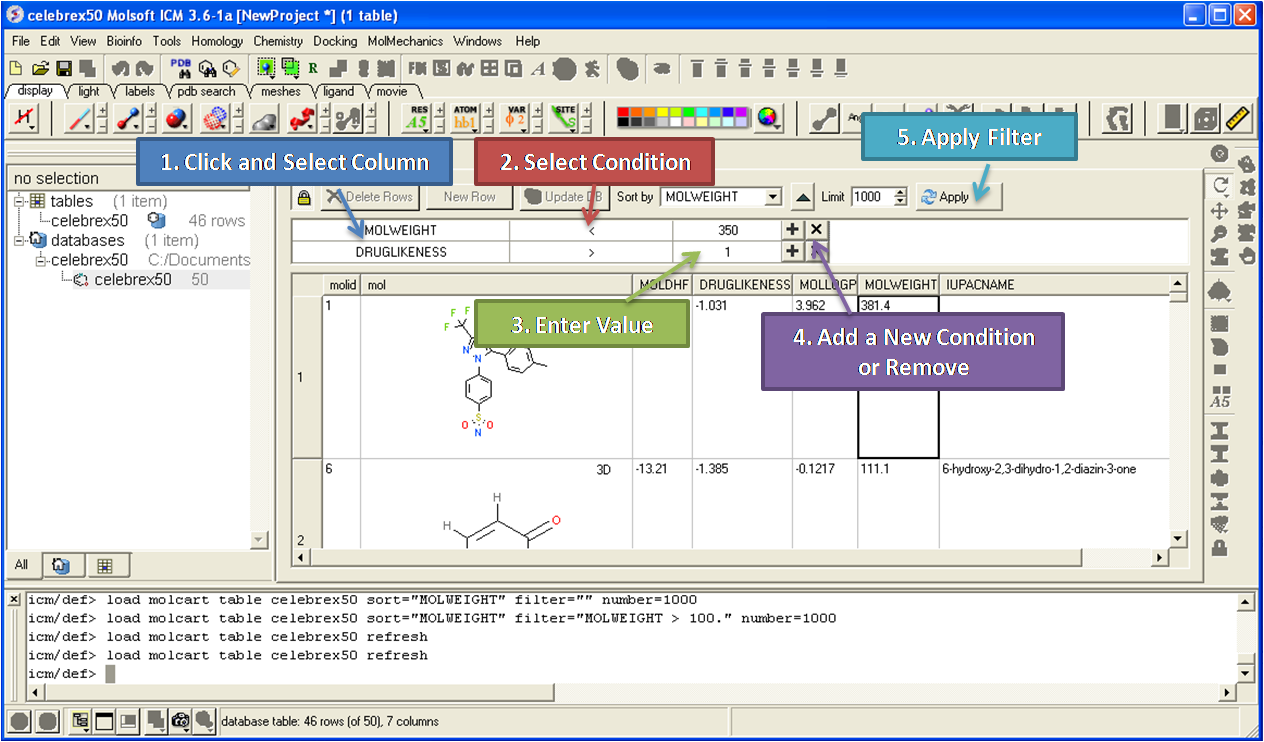
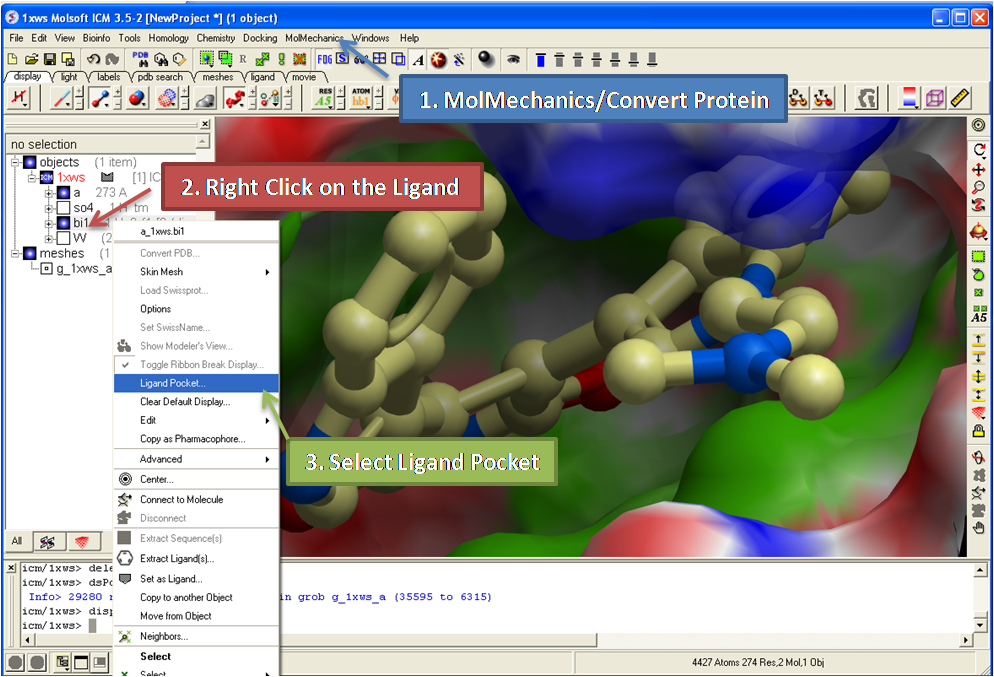


 On January 25th-27th 2006, a 3-day ICM Workshop was held at Molsoft's La Jolla
offices. The first two days of the workshop covered all aspects of protein
structure and drug discovery including sequence analysis, protein modeling,
small molecule docking, protein-protein docking, cheminformatics and QSAR. The
third day was dedicated to advanced ICM training including the ICM command
language, scripting, loop modeling and flexible receptor docking. The course was conducted by Prof. Ruben Abagyan (The Scripps Research Institute) and
Dr. Maxim Totrov (Principal Scientist - Molsoft).
On January 25th-27th 2006, a 3-day ICM Workshop was held at Molsoft's La Jolla
offices. The first two days of the workshop covered all aspects of protein
structure and drug discovery including sequence analysis, protein modeling,
small molecule docking, protein-protein docking, cheminformatics and QSAR. The
third day was dedicated to advanced ICM training including the ICM command
language, scripting, loop modeling and flexible receptor docking. The course was conducted by Prof. Ruben Abagyan (The Scripps Research Institute) and
Dr. Maxim Totrov (Principal Scientist - Molsoft).
 The new version, ICM 3.0, was introduced at the course and was well accepted as
an extremely user-friendly interface by the attendees. This version, which also
has stereo viewing mode that works with VRex glasses on Linux and Windows
platforms, was welcomed by all the participants as an efficient and low cost
alternative to existing stereo viewing tools.
The new version, ICM 3.0, was introduced at the course and was well accepted as
an extremely user-friendly interface by the attendees. This version, which also
has stereo viewing mode that works with VRex glasses on Linux and Windows
platforms, was welcomed by all the participants as an efficient and low cost
alternative to existing stereo viewing tools.

 Molsoft is pleased to announce its participation at the Protein Society's 16th
Annual Symposium held at the Marriott in San Diego from August 17-21, 2002.
Attendance was estimated at around 1300. Molsoft's exhibit was designed to
inform Protein Society attendees about the latest technology available from
Molsoft including its recent version of a new graphical interface which enables
Molsoft's ICM software easier to use than ever before.
Molsoft is pleased to announce its participation at the Protein Society's 16th
Annual Symposium held at the Marriott in San Diego from August 17-21, 2002.
Attendance was estimated at around 1300. Molsoft's exhibit was designed to
inform Protein Society attendees about the latest technology available from
Molsoft including its recent version of a new graphical interface which enables
Molsoft's ICM software easier to use than ever before.




{kind=link}