| Prev | 2.8 Selections | Next |
[ os_ | ms_ | rs_ | as_ | vs_ | selfunctions | selranges ]
Let us imagine that we decided to compare two structures deposited in the PDB. We will read both entries in the ICM shell, and define the following levels or organization. Each entry will form an object, each object will contain one or several molecules, protein molecules will naturally contain amino acid residues and residues will consist of atoms. Now, in the superimpose command, we will need to specify, or select, the molecules, residues or atoms which should be superimposed. The ICM shell language has a flexible way of selecting subsets of atoms, amino-acid residues, molecules, objects, as well as torsion angles and other internal geometrical parameters of molecules. Most of the ICM commands and functions dealing with molecules, for example, display, delete, minimize, etc., will operate on an arbitrary selection. What does a selection look like? For example, selection a_2./2:14/c* selects carbon atoms of residues from 2 to 14 of the second object. The general syntax of a selection is the following:prefix _ [ object(s) . ] molecule(s) / residue(s) / atom(s) or variable(s)
The object section including the dot (e.g. 1crn. ) may be omitted. In this case the selection will be performed in the current object.
There can be as many as five sections separated by _ . / and /,
Examples:
a_2ins.a,b/lys,arg/ca,cb,n* # atom selection, '*' - any string a_2ins.a,b/2:10/n,ca,c # atom selection v_crn./lys,arg/phi,PSI # variable selection(Note use of PSI torsion in the last example.)
Storing selections in named variables.
Selections can be assigned to a variable (e.g. x = a_//c* ) and can be combined in an expression by logical and ( & ) or logical or ( | ), e.g. ( a_//n* & a_//ca ).
!- Selection Types
Three prefix types: a_ v_ and V_ . The Prefix defines one of the three selection types:
- atoms, residues, molecules and objects ( a_.. )
- free variables ( v_.. )
- all variables ( V_.. )
| abbr. | selection name | example |
|---|---|---|
| os_ | object selection | a_ ; a_1. ; a_1crn. ; a_*. |
| ms_ | molecule selection | a_1.2 ; a_a,b ; a_*.* |
| rs_ | residue selection | a_/3:9 ; a_/* ; a_/"GKS" |
| as_ | atom selection | a_1.2//ca,c,n ; a_//c* |
| abbr. | selection name | example |
| vs_ | selection from free internal variable | v_ ; v_1. ; v_1.2//x* ; v_2//?vt* |
| Vs_ | selection from all internal coordinates | V_ ; V_1. ; V_1crn.//!phi,psi,omg |
A selection can also be assigned to a named variable:
Example:
aa = a_//ca,c,n # the backbone show aa
The object and molecule sections are separated by a period, all other sections are separated by slashes. Inside each section, arguments in a list are separated by comma (,) while ranges are separated by colon ( from:to ).
!- Selection levels
There are four principal levels of selection: object selection, molecular selection, residue selection and atom or variable selection. The level is defined by the "lowest" section explicitly specified in a selection (e.g. a_1.1/2:4 is a residue level selection, while a_//ca is an atom selection). These selections are referred to as os_ ms_ rs_ as_ or vs_ , respectively. If selection level is not important or the level is the lowest one (atoms or variables), selections are referred to as as_ or vs_.
The selection level of the interactive graphics selections is controlled by the GRAPHICS.selectionLevel preference. To change it from the command line, assign this variable to an appropriate level, e.g. GRAPHICS.selectionLevel="atom" .
Selection levels can be changed from the GUI interface, by changing the selection level
!- Examples
Examples of different selection levels (note that object and molecule names are arbitrary):
a_1,3. a_mod*. a_*. a_"*benz?n*". # object selections a_3.mol1 a_zinc a_$molNum a_*.* # molecule selections a_/3:29,as?,ala a_/* a_*./"VHC?[!W]A" # residue selections a//h?,c* a_//T v_//phi,psi # atom or variable selectionsFor example, a_1,3. is an object selection, and a_/ala is a residue selection.
Each section may contain a negation symbol ! in the beginning. It selects all, but the specified. You can only use the negation symbol in the first position of a section and the negation will always apply to the whole section. For example, a_/!ala,gly is right, while a_/ala,!gly is wrong.
If object section together with the separating period is skipped, selection addresses the current object rather than all objects.
!- Select by number, range, name or pattern
Matching. Objects, molecules, residues, atoms and variables may be referred to by their names. Objects and molecules can be additionally referred to by their sequential numbers (e.g. a_1.2). To select by a numerical name, use backslash before the name, e.g. a_\123 . Metacharacters, such as * ? [], can also be used for pattern matching (e.g. v_//?vt*).
Full syntax. A complete description of selection syntax for each level is as follows:
2.8.1 Object selection | [Top] |
| a_ name . ( a_1crn. , note the dot at the end ) | |
| a_ namePattern. ( a_1c?n. ) | |
| a_ relNumber. ( a_2. means the second object) | |
| a_ num1:num2. ( a_2:5. range from object 2 to object 5 ) | |
| a_ | the current object, it is a special case. |
| a_ " commentPattern ". | select by pattern matching in the object comment field. |
| a_ICM. | objects of ICM type ( a_!ICM. - non-ICM objects) |
| a_CATRACE. | objects of "Ca-trace" type |
Example:
read object s_icmhome+"all" show a_ # the current object show a_1,2:3. show a_s1?. show a_"*Th[iy]o*".//!h*
2.8.2 Molecule selection | [Top] |
[ wateraccess ]
| a_obj.mol | in specified object(s), |
| a_mol | in the current object or |
| a_*.mol | in any object |
by name:
a_s_name e.g. a_m2 or a_1.m2 in the current ( a_ ), or the first ( a_1 ) object, respectively. ( Note that there is no dot at the end ). If the name starts with a digit or one of the reserved one-letter types (see below), add backslash before the digit, e.g. a_\123 , a_\A .
by pattern
a_s_namePattern ( a_w* - all water molecules in the current object)
by number(s)
a_number ( a_2 , a_3.2,4,7 ) - relative number of molecule(s)
by range(s)
a_num1:num2 ( a_2:5 , a_2:5,10:12 ) - number range
by chemical formula (F):
a_Fformula1,Fformula2..
the chemical formula must be the same as the one returned by the ICM String( ms_ ) function without hydrogens, e.g.
read pdb "1abe" show a_FC505 # selects 2 arabinose molecules String( a_2//!h* ) C5O5
by special symbol for types of molecules:
a_specialSymbol[,specialSymbol2..]
- A peptides and proteins
- B molecules included in biological unit
- C
select by Chain, e.g. a_1.Cabc - H hetatm, usually ligands and water molecules
- N nucleic acids
- Q molecules which have a seQuence linked to them
- S sugars
- M Multiple sequence alignment linked to molecule exists
- L lipids
- W water including deuterated water (dod)
- U unknown (miscellanea)
Note that if a molecule name coincides with any of the above characters ( i.e. "ACHLMNQRSTUW" ), ICM gives preference to the type selection. To select by molecule name, use backslash (e.g. a_1.\A for chain named "A" )
Examples:
nice "1dnk" # one peptide, two dna chains and other mols a_A # the peptide a_N # the two DNA chains a_A,N # the peptide and the DNA chains rename a_1 "A" a_\A # chain NAMED "A" read pdb "2ins" delete a_W
Some special cases:
a_* # all molecules in the current object a_a # molecule 'a' in the current object a_.a # molecules 'a' in all objects a_*.a # the same as a_.a
selecting water molecules from pdb-files by their 'residue-field' number.
Water molecules in PDB files are numbered and the numbers are stored in the residue field. For consistency, we convert these numbers into residue numbers. At the same time the names of water molecules are built sequentially like this: w1,w2,w3 . This way one can use both sequential numbering via molecule names and PDB-file numbering via residue numbers.
read pdb "1sri" show a_w12:w15 # by molecule name, sequential numbering show a_w*/719:721 # by original pdb number
converting any selection to molecules with the Mol function
Selection of any level, e.g. atoms, residues, and objects can be converted to molecules with the Mol ( selection ) function. Example:
Mol(Sphere(a_zinc a_1,2 8.)) # Sphere returns atoms
2.8.3 Residue selection | [Top] |
| a_obj.mol/res | complete specification, (e.g. a_*.*/14:19 or a_2.3/ala). |
| a_mol/res | the current object and the specified molecules, (e.g. a_w*/* ) |
| a_/res | all molecules of the current object, (e.g. a_/23:25) |
Residue field specifications (for all molecules in the current object).
by name:
a_/resName ( e.g. a_/his , or a_/\001 - here we had to start with a backslash becase the residue name looked like a number)
by pattern:
a_/resNamePattern ( e.g. a_/as? - asn or asp). A useful tip for DNA or RNA selections. Quite often bases are modified. To select A,T,G,C,U and their modifications, use a_/??a or a_/??t or a_/??g or a_/??c or a_/??u, respectively.
by residue number(s):
a_/numChar ( a_/3 or a_/15A ) - PDB residue number may contain additional characters.
by residue range(s):
a_/numChar1:numChar2 ( a_/4:15,20:25 ) - reference residue number range
by amino acid sequence pattern:
a_/"seqPattern" ( a_/"G?GTE" ) - selects the fragment with matching aminoacid sequence.
Example selecting all residues preceding prolines (the first expression selects dipeptides with the second proline, the second one excludes prolines):
show a_/"?P" & a_/!pro*
by special symbols and expressions
by residue type
a_/A - residues of "Amino" type (N- and C-termini have different type) displayed residues
a_/D - displayed residues in the ribbon representation only
a_/DD - displayed residues in which either ribbon or some atoms are displayed
residues identical to their homology target residues
a_/I - if atoms of one molecular object are tethered to atoms of another object, selection a_/I shows those tethered residues (i.e. they contain tethered atoms) which have identical names to the residues to which they have been tethered.
by absolute number
a_/N absNumber ( a_/N15 ) - absolute number (all residues
of all objects are numbered sequentially starting from one.) by secondary structure
a_/S sec_struct_chars - residues with certain secondary structure (e.g. a_/SH - only helices; a_/SEH - sheets and helices; a_/S_ - only coil)
terminal residues (like N-terminal, C-terminal, and DNA 5' and 3' termini ) a_/T
by alignment consensus
a_/C resConservationCode - selects residues according to the consensus of the alignment linked to a molecule. The symbols can be combined, e.g. a_/CYnh for conserved tyrosines, negatively-charged residues and hydrophobics. Possible codes:
- A , C ... - particular conserved amino acid types (one-letter code)
- X - all absolutely conserved residues
- h - conserved hydrophobic residues (#)
- s - conserved small residues (^)
- p - conserved polar residues (~)
- o - conserved positive residues (+)
- n - conserved negative residues (-)
- a - conserved aromatic residues (%)
- x - not conserved but in the ungapped block (.)
- g - gap in one of the sequences of the alignment (' ')
by functional features
a_/F[SiteChars] or a_/F"siteID"
residue selection by the one-letter site type or the site ID, respectively. Letter F refers to the word feature as in the FT (feature table) field of Swissprot entries. The types along with their one-letter codes are listed in the glossary site entry. The default string, the a_/F selection, is defined by the SITE.defSelect string (you may redefine it), which defines important local features such as binding sites as opposed to domain-type sites such as signal peptides, zinc fingers and other protein domains. The PDB entries do not comply with the standard SWISSPROT site definitions, such as ACT_SITE BINDING etc., and are assigned by the user type F (selection a_/FF ).
Example:
nice "1as6" show site color ribbon a_/F magenta show a_/FF show a_/F"cu3" # select only site named cu3 show a_/F"MUTAGEN" # sites so defined in Swissprot set site a_1.1 "FT SITE 15 15 My favourite residue"
converting selections to residue level: The Residue( selection ) will convert any selection of higher level or lower level to the residue level. Example
a_/SH & a_/pro # a proline in a helix Res(Sphere(a_/pro 2.)) # expand to the neighboring residues
2.8.4 Atom selection | [Top] |
[ as_alter | as_bycode | as_onscreen | as_bygrad | as_bytether | as_bytetherdest ]
( a_//atoms ):by name
a_//name ( a_.//ca , ca is a usual name for alpha carbon )
by name pattern
a_//namePattern ( a_.//c* for all carbons )
by special symbols and expressions
alternative atom positions in X-ray structures
a_//A alterCharacter - select alternative positions of the specified type (e.g. read pdb "1cbn" ; show a_//Ab ). See also the set comment "A" as_ command.
by atom code
a_//CatomCodeNum[:atomCodeNum2] - select by atom code as described in the icm.cod file, e.g. a_//C2,C4 selects aromatic and methylene hydrogens, a_//C2:15 selects codes from 2 to 15
a_//MatomMmffCodeNum[:atomCodeMmffNum2] - by mmff code
displayed atoms
* a_/D[displayTypes] - Displayed atoms (e.g. a_//D for all displayed atoms, or a_//DWC for wire or cpk). The following graphical types can be selected:
- A - labelled atoms
- B - ball
- C - cpk
- D - displayed atoms or atoms in displayed residues
- S - skin
- W - wire
- X - xstick (i.e. ball or stick)
- no arguments - any graphical representation
Special named selections: as_graph graphically selected atoms:
as_graph selection contains graphically selected objects, molecules, residues, or atoms The level of selection depends on the GRAPHICS.selectionLevel preference. The level can be changed from the GUI interface or from command line.
strained atoms (atoms with high energy gradient)
a_//G - strained atoms (Gradient vector longer than selectMinGrad) You can also use the display gradient command.
Example:
buildpep "his trp trp" display randomize v_//phi,psi selectMinGrad = 100. show energy display a_//G ball display gradient
hydrophobic atoms a_//H
aromatic atoms a_//R It selects heavy atoms connected by aromatic bonds and hydrogens attached to them. Example:
buildpep "HWYP" display skin color skin a_//R magenta
tethered atoms
a_//T - Tethered atoms (see also a_//Z - tether destination atoms)
tether-target atoms
a_//Z - Tether destination/target atoms (see also a_//T - tethered atoms)
chiral atoms a_//X[0123RLB] - chiral atoms. Each atom has two bits characterizing its chiral properties. If the two bits are presented as an integer, the chiral number has the following values:
- zero - a non-chiral center
- 1 - a left topoisomer (L)
- 2 - a right topoisomer (R)
- 3 - a racemic mixture of both isomers (B)
by absolute number
a_//absNumber - absolute number (all atoms of all objects are numbered sequentially starting from one)
converting to atom level: The Atom ( selection ) will convert any selection of higher level to the atom level.
2.8.5 Free and all variables (v_ and V_) | [Top] |
|
The v_selection selects free variables in molecular objects of ICM-type.
The main types of internal coordinates , or geometrical variables, are shown below: |
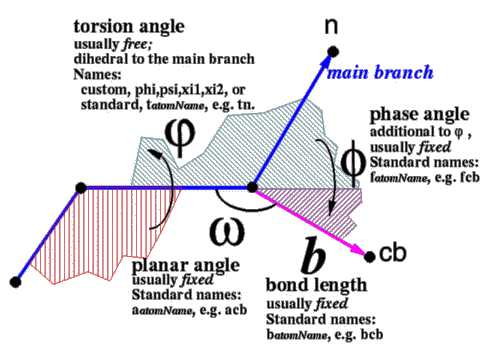
|
The position of each atom branch is determined by the positions of the preceding atoms and three parameters: dihedral angle, planar angle and bond length. The dihedral angle for the main branch atom is the torsion angle itself, while for the secondary branch atoms the dihedral angle consists of the torsion angle plus the phase angle. The default fixation is given in the ICM-residue library and can be changed by fix and unfix commands. Individual free variables can be rotated interactively with Ctrl-LeftMB-Atom-Click and drag. A vselection can also be assigned to a named variable:
Example:
aa = v_//phi,psi # the backbone torsions unfix only aa unfix only v_/10:15/phi,psi
V_ : selecting among all internal coordinates
Finally, the V_ selection selects both free and fixed variables in molecular objects of ICM-type. You always need this type of selection in the unfix command. It makes no sense to unfix variables which are free already.
Here is a list of variable selection specifications:
by name:
v_//name ( v_//phi )
by name pattern:
v_//namePattern ( v_//x* ) use asterisk * for any string, and question mark ? for any character. Example: v_//?vt* selects the 6 " virtual" variables defining rigid body rotation and translation.
torsion variables
v_//TtorsionCodeNum[:torsionCodeNum2] - select by torsion angle code as described in the icm.tot file, e.g. v_//T11 selects the amide group torsion angle v_//T10:15 selects torsion codes from 10 to 15
angles (planar angle variables)
v_//AangleCodeNum[:angleCodeNum2] - select by planar angle code as described in the icm.bbt file.
bond length variables
v_//BbondCodeNum[:bondCodeNum2] - select by bond length code as described in the icm.bst file.
Psi torsions not shifted to the next residue
v_//PSI - psi torsion angle which belongs to the residue you would expect. The reason for this definition is that from ICM point of the psi backbone torsion with rotation axis between Ca and C of residue i belongs to N-atom of the next residue i+1 because N is the first atom this torsion angle moves. E.g., v_/3/phi,psi selection will contain the psi from residue 2 and then phi from residue 3. The definition PSI allows you to use the conventional attribution of angles, e.g. v_/3/phi,PSI is a pair of angles with axes around Ca atom or residue 3. Important. However, note that if you use selection expressions like
v_//phi,PSI & a_/2,3 it will not work (in contrast to a_/2,3/phi,PSI ) and you will have to use the Next function.
Example:
vPhi = v_/3/phi vPsi = v_/3/PSI # BUT !!! vPhi = v_//phi* & a_/3 vPsi = v_//PSI & Next( a_/3 )
methyl group torsions
v_//M - torsion angles rotating Methyl-type terminal hydrogens (excluding polar hydrogen)
polar hydrogen torsions
v_//P - torsion angles rotating Polar hydrogens (e.g. hydroxyl group)
essential (non-hydrogen) torsions:
v_//H - side chain torsion angles rotating "Heavy" atoms
standard set of free torsions (excludes rings)
v_//S - all "Standard" free torsion angles as defined in the icm.tot file.
Note that v_//M, v_//P, and v_//H do not overlap, they are mutually exclusive. v_//S contains v_//M, v_//P, and v_//H as well as other standard torsion angles.
phase angles
v_//F - select all phase angles (usually they are fixed, so use V_//F )
V_//FC - select phase angles related to the chiral centers (see set chiral and montecarlo chiral )
all torsion angles
v_//T - select all free torsion angles, V_//T for all torsion angles including the fixed ones.
2.8.6 Functions returning selections | [Top] |
- Acc - select solvent-accessible atom/residues.
- Atom - convert to the atom selection
- Deletion - residues deleted according to the alignment
- Insertion - residues inserted according the alignment
- Mol - convert to the molecule selection
- Next - extract the next atom
- Obj - convert to the object selection
- Res - convert to the residue selection
- Sphere - expand a selection by r_radius or 5Ĺ.
- Select - selection of atoms according to their coordinates, bfactors, or other properties
Substituting ICM-shell variables into a selection. You can insert the value of an integer or string ICM-shell variable anywhere inside your selection by using a $ (dollar sign) prefix. (Note, this is a general ICM-shell substitution mechanism).
Examples:
selstr="!w*/14:19" # a string constant display a_$selstr
Logical operations. You can also assign selection to a variable, (i.e.: backbone=a_//ca,c,n ) combine several selections using logical operators (example: show a_/3:6 & backbone ) .
2.8.7 Finding contiguous residue ranges with the String function | [Top] |
read pdb "1dkf"
rrange = String( a_/"?P?" ) # the result would look like "a_a.b/5:7,30:32"
rg = Split(rrange,"/,|") # split into sarray with {"a_a.b","5:7","30:32"}
# bar (|) helps with multiple chains
okrg={""}
k=0 # counter for good residue triplets with HHH and ?P?
for i=2,Nof(rg)
if Nof(Split(rg[i],":")) != 2 continue # ignore molecular names
if Sstructure( a_/$rg[i] ) == "HHH" then # compare with ss-pattern
k = k+1
okrg[k] = rg[i]
endif
endfor
# now ok-ranges are stored in okrg string array e.g. {"5:7"}
# to use them Sum(okrg,",")
| Prev molecule | Home Up | Next arithmetics |