| Prev | ICM User's Guide 16.4 Build Prediction Model | Next |
Structure-Activity Relationship (SAR) is a process by which the activity of a molecule is related to its molecular structure. If a significant ammount of structural and activity data is available a model can be made which can be used to predict the activity of a molecule or series of molecules.
In ICM SAR is undertaken using the Learn and Predict tools in a Molecular Table.
Learn
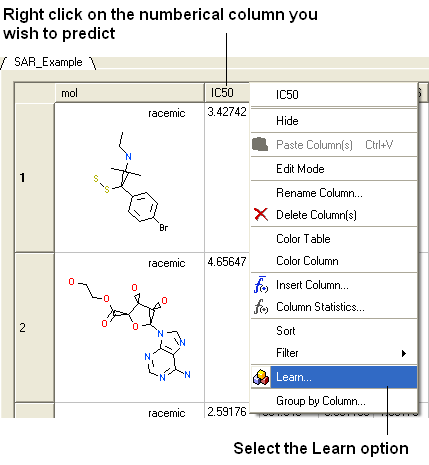
Step 1: Select the column you wish to predict and then Tools/Table/Learn or use the right click option shown below.
Step 2: Fill in the Learn options as shown below.
- Enter the name of table with which you want to perform the predictions. You may locate your table from the drop down arrow menu.
- Select the column from which you wish to learn. Use the drop down arrow to select.
| NOTE If the table does not contain any numeric (integer or real) columns, there is nothing to predict, so the "Learn" button will be disabled. |
- Enter a name for the learn model.
- Select which regression method you wish to use from the drop down menu. See the theory section to determine which method and parameters to use.
- Select which columns (descriptors) of your table you wish to use to 'learn'.
- If you are using chemical descriptors to produce your model select the maximal chain length.
- Select the number of cross-validation groups you wish to use or selected rows can be used for cross validation. The number of iterations will impact the speed of the calculation. 5 is the default number of groups but 2 would be the least rigorous and selecting the 'Leave-1-out' would be the most rigorous calculation.
- Click on the learn button and a table summarizing your model will be displayed as shown below.
| Prev Annotate | Home Up | Next Predict |