| Prev | ICM User's Guide 22.16 Docking Examples | Next |
[ Re-Dock Biotin to the Streptavidin Receptor | Re-Dock an Inhibitor to Ricin Crystal Structure ]
22.16.1 Re-Dock Biotin to the Streptavidin Receptor |
Objective
To dock biotin into the streptavidin receptor.
Instructions
- Docking/Set Project Name (BIOTIN)
- Docking/Load Example (streptavidin complexed with haba)
- Select biotin in workspace window and right click and select neighbors.
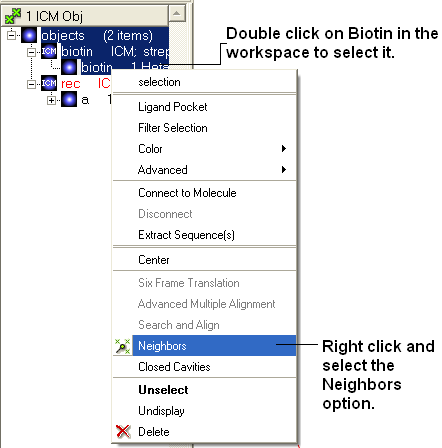
- Select atoms on other objects in a 5 A radius.
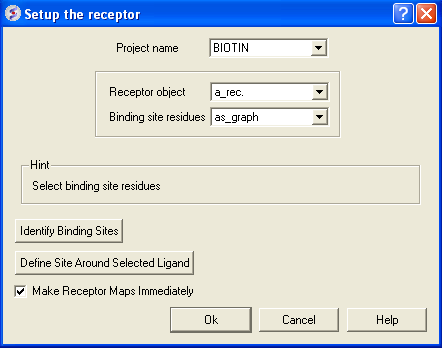
- Docking/Receptor setup (Receptor molecules: a_rec.a)
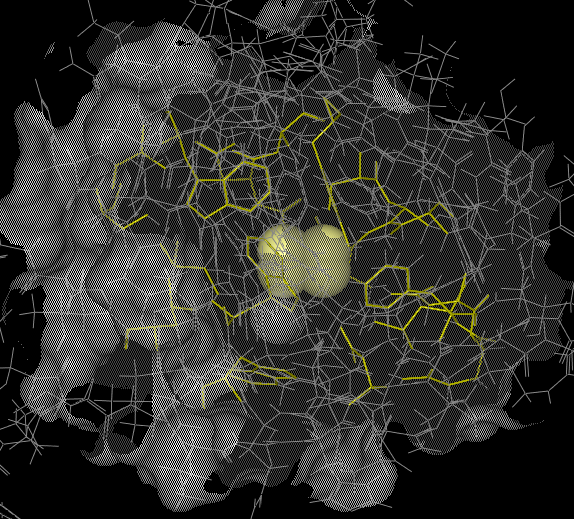
The binding pocket should be displayed something like as shown below:
In the terminal window instructions will be given telling you how to alter the initial starting position of the ligand and how to change the size of the box in which the maps will be generated.
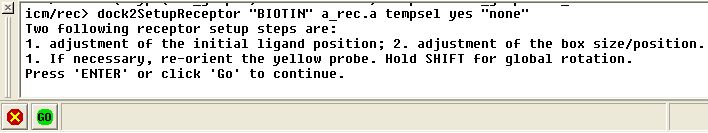
Adjust box size / probe position and press return or use the green GO button in the bottom left hand corner.
Now let us set up the ligand and start the simulation:
- Docking/Interactive Docking/From Loaded ICM object (Ligand molecule: a_biotin.biotin)
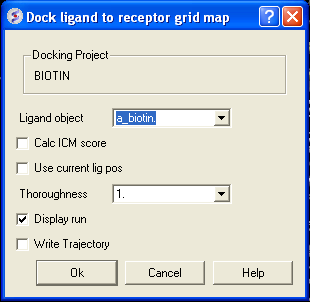
<>
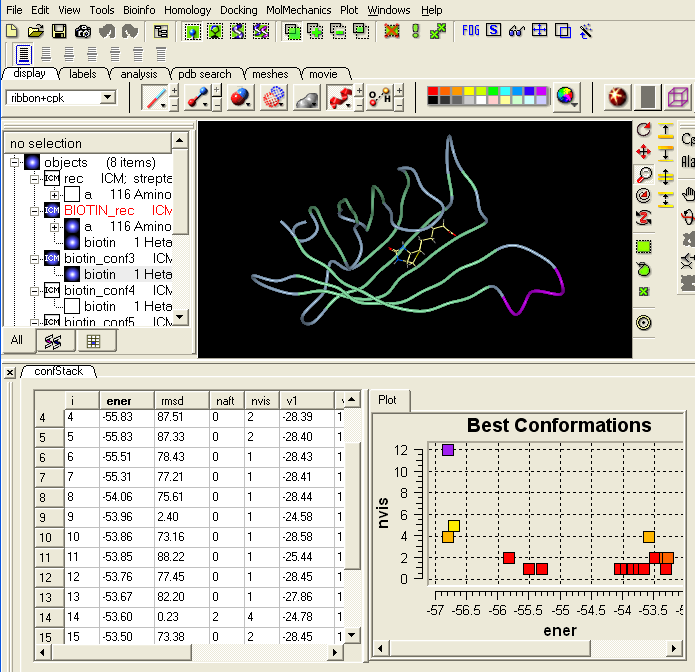
Once the simualtion has finished select
- MolMechanics > View Stack
- Double click on each element of the stack and the ligand will be displayed in the graphical display. Each ligand position can be displayed or undisplayed using the ICM workspace.
22.16.2 Re-Dock an Inhibitor to Ricin Crystal Structure |
Objective
To re-dock a ricin inhibitor into the ricin crystal structure (1br6).
Instructions
- Select the Pdb Search tab and type ricin followed by the 'Enter" key.
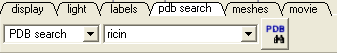
- A table as shown below will be displayed. Double-click on 1br6
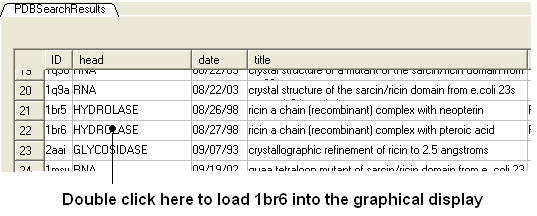
- Right click on 1br6 in the ICM Workspace and select Clone and call it ligand.
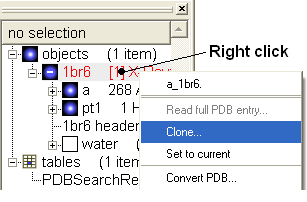
- From the first object delete the small molecule (pt1) by right clicking on it in the ICM workspace. From the clone, delete the receptor (a), and water
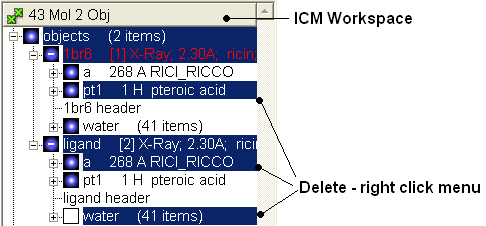
- MolMechanics >ICM-Convert >Protein (Object: a_1br6., replace the original)
- MolMechanics >ICM-Convert >Chemical (Object: a_ligand. )
- Docking > Set Project Name (RICIN)
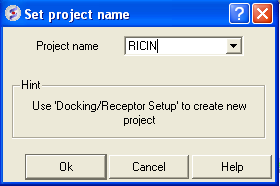
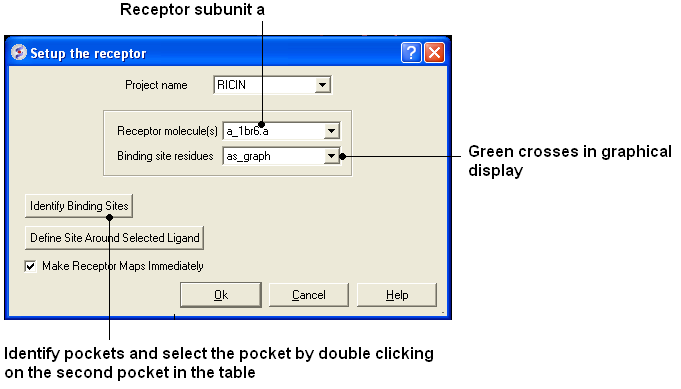
- Docking > Receptor Setup. Fill in the boxes as shown below and select the Identify binding sites button Select second pocket in the table, Click OK. Press Enter or click on the "GO" button. In this example there is no need to change the box size or probe position.
- Docking/Review/Adjust Ligand/Box (Box can be resized)
- Docking/Make receptor maps
- Docking > Interactive docking > Loaded Ligand (Ligand: a_ligand. )
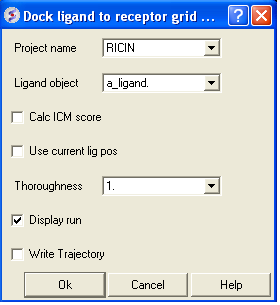
| NOTE: The simulation may take longer if you are displaying the ligand during docking. |
Once the docking has finished you can display the best energy solution
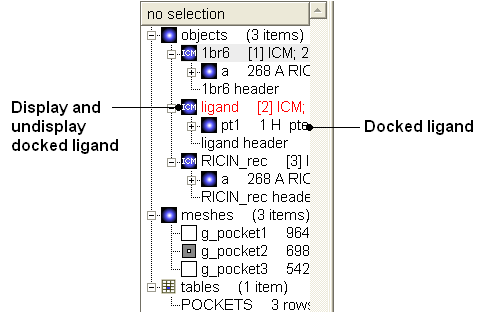
Compare with crystal structure
- Read in 1br6 and display ligand. Note docking pose for this example can be improved by including the water molecules w37 and w41 which are inside the binding pocket. To include water molecules remember not to delete water in the pdb conversion to object and type the following in the receptor setup stage a_1br6.a,w37,w41
- You can drag and drop the docked ligand into the receptor inside the ICM Workspace resulting in a single complex (see FAQ section "How can I merge two separate objects into one?")
| Prev Predict | Home Up | Next VS Examples |