| Prev | ICM User's Guide 22.17 Virtual Screening Examples | Next |
[ VLS - Ricin | VLS - Cyclooxygenase ]
22.17.1 Virtual Ligand Screening to Ricin Receptor |
Objective
To perform virtual screening into the ricin receptor.
Instructions
- Docking> Set Project (select RICIN) see previous lesson Re-Dock an Inhibitor to Ricin Crystal Structure.
- Docking> Tools> Index Mol/Mol2 file/database (Input file . select ricinLigands2D.sdf, )
- Docking> Ligand Setup> From Database (select mydb.inx, check .mol., .build hydrogens. .assign charges. and .2D to 3D. convert)
- Docking > Run Docking Batch
- Docking > Make Hit List (select import 2D from DB)
- Browse HITLIST table
22.17.2 Virtual Ligand Screening to Cyclooxygenase |
Objective: To dock indomethacin and perform virtual screening of a database of COX inhibitors into the Cyclooxygenase receptor.
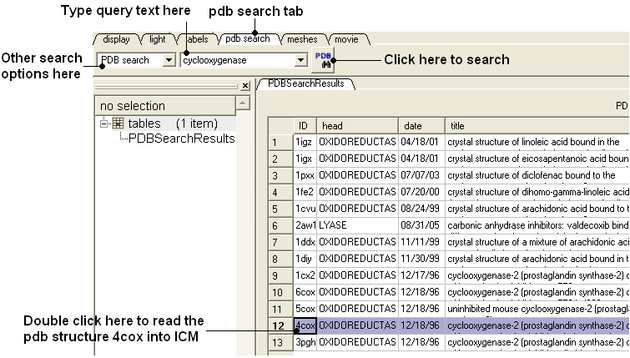
Retrieve the Cyclooxygenase receptor 4cox from the protein databank.
- Select the PDB Search tab and type 'Cyclooxygenase' and hit the PDB button.
- Find the pdb entry 4cox in the table and double click on the row to load it into ICM.
- Right click on the pdb file 4cox in the ICM Workspace and select clone - use the default options and select OK.
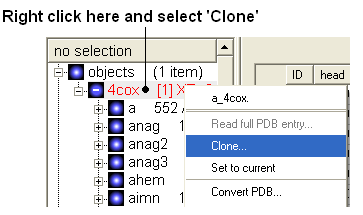
- Enter object name ligand.
- From the first object (**4cox) delete everything except for the first molecule 'a'
- From the second object (**ligand) delete everything except for aimn
| NOTE: To delete molecules you need to select them in the ICM Workspace and then right click and select delete. A range of molecules can be selected by clicking on one and whilst holding the Shift button click on the last molecule. Non-contiguous selections can be made using the Ctrl button. |
- The ICM Workspace should now look something like this:
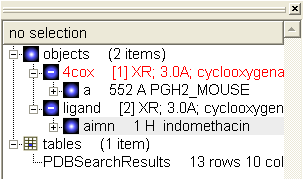
Converting the Ligand and Receptor into an ICM Object.
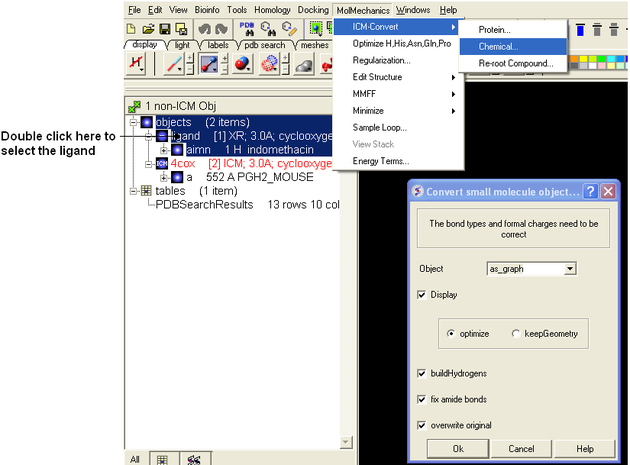
- Double click on the first object 4COX and select MolMechanics/ICM-Convert/Protein - choose the options shown below
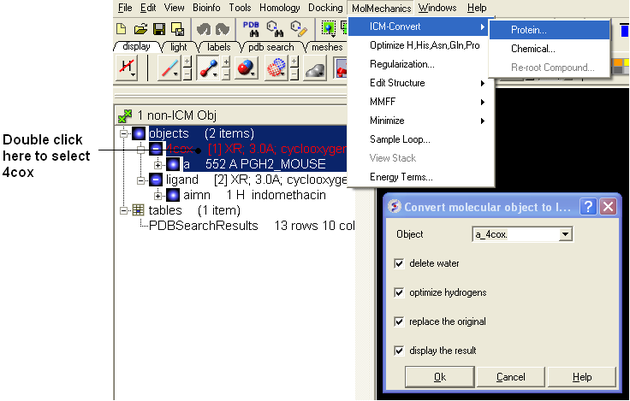
- Double click on the second object ligand and select MolMechanics/ICM-Convert/Chemical - choose the options shown below
Setting up the docking experiment
- Docking/Set Project enter COX2
- Select 4cox in the ICM Workspace (double click in the ICM Workspace on 4cox - should be highlighted blue in the ICM Workspace and green crosses in the graphical display).
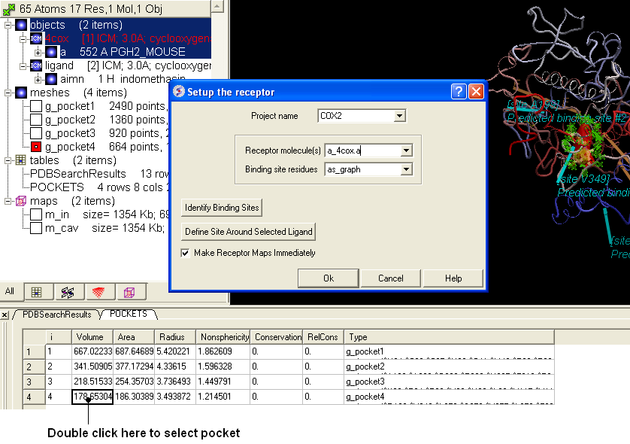
- Docking/Receptor Setup and select the Identify Binding Sites button.
- Select the 4th pocket in the POCKETS table and you should see green selection crosses around the binding site as shown below.
- Fill in the Setup the receptor windows as shown below and press OK.
- Press the *GO button in the button left hand corner of the GUI twice. In this example there is no need to change the position of the probe or the box.
- Docking/Make Receptor Maps and press the OK button.
- Docking/Interactive Docking/Loaded Ligand - (see below)
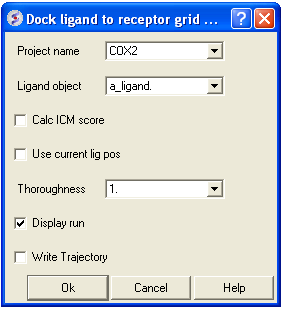
- The ligand will be seen on the screen sampling the pocket.
- The final docked ligand pose will be displayed and is in the ICM Workspace
| To compare the docked pose with the crystal structure - we need to rename the first object 4cox to 4cox_receptor (or just delete the first object) and then double click on 4cox in the PDBSearchResults table we used earlier. |
Now we will dock Vioxx into the Cox receptor
- In the ICM distribution (cd $ICMHOME or C:Project Files/MolSoft LLC) you can find a file called vioxx.sdf. If you cannot find this file please E mail support@molsoft.com and we can send it to you.
- File/Open and find the vioxx.sdf file
- Docking/Interactive Docking/Mol Table Ligand
- Compare the docked pose of Vioxx with the crystal structure 1cx2 (pdb search 1cx2)
Now let us perform a virtual screen of a database of COX inhibitors
- Docking/Set Project enter COX2
- Docking/Tools/Index Mol/Mol2/ file/database and select celebrex50.sdf . If you cannot find this file please E mail support@molsoft.com and we can send it to you (cd $ICMHOME or C:Project Files/MolSoft LLC).
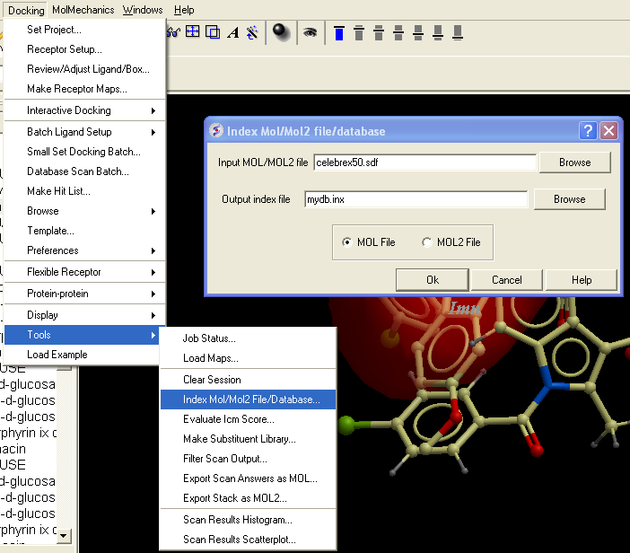
- Docking/Ligand Setup/From Database and select mydb.inx - check mol,build hydrogens, assign charges and 2D to 3D convert
- Docking/Run Docking Batch
You can check up on the progress of the docking by selecting Windows/Background Jobs. A messsage will be displayed on the screen when the docking is finished.
- Docking/Make Hitlist select import 2D from DB
- Browse Hitlist table to view docked complexes.
| Prev Re-Dock an Inhibitor to Ricin Crystal Structure | Home Up | Next Docking a Markush Generated Library |