| Prev | ICM User's Guide 22.19 Multiple Receptor Conformation Ensemble Docking Example | Next |
Background
Here we will demonstrate multiple receptor conformation docking in ICM using Aldose Reductase as an example. Aldose Reductase has a flexible loop in the ligand binding pocket vicinity which enables a variety of inhibitors to bind therefore in order to identify these ligands via docking it is necessary to sample the conformations of this loop and dock to an ensemble of structures. Step 1 to Step 3 takes you through the standard re-docking procedure and highlights the accuracy of re-docking a ligand to the receptor conformation from which it was crystallized. Step 4 demonstrates that a single conformation of the Aldose Reductase receptor cannot account for the binding of all known aldose reductase inhibitors. Step 5 outlines the steps for sampling the conformation of the flexible loop regions and generating a series of conformations (stack). Step 6 is the multiple receptor docking stage (4D docking) and the results are viewed in a hitlist with an additional column which reports the receptor conformation that the highest scoring ligand is bound to.
Step1: Prepare the Receptor
- Load PDB file 1pwm. You can search for a PDB file using the PDB Search tab.
- Delete the chlorine atom. This can be done by right clicking on the chlorine in the ICM Workspace and selecting delete.
- Move the ligand (fid) out of the receptor. You can do this by right clicking on the ligand (fid) in the ICM Workspace and select Move From Object.
- Convert 1pwm into an ICM object. MolMechanics/ICM-Convert/Protein
- Rename the small molecule (fid) object "ligand" and the receptor object "receptor". You can rename an object by right clicking on the name of the object in the ICM Workspace and select rename.
Step2: Setup the Docking Project
- Docking/Set Project and enter the project name ALDR
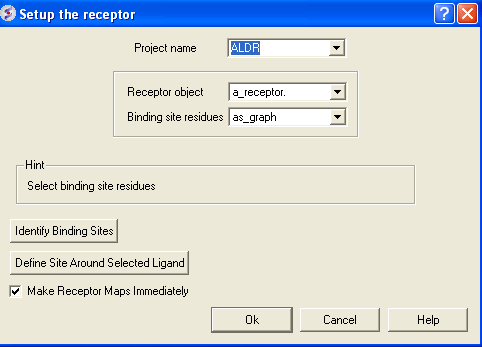
- Docking/Setup Recptor Enter the data as shown below. Double click on the ligand in the ICM Workspace and then select the Define Site Around Selected Ligand button (you should see green crosses surrounding the ligand on the receptor).
- Click OK and then the green GO button twice and wait for the maps to be generated.
Step 3: Optional: Re-Docking the X-ray Ligand
- Drag the ligand out of the pocket so that we can re-dock it. You can do this using the CONNECT option. Right click on the ligand and select Connect to Object and then use the middle mouse button to drag the ligand. Once you have moved the ligand press the escape button.
- Convert ligand into an ICM object. MolMechanics/ICM-Convert/Chemical - select or enter a_ligand.
- Docking/Setup Batch Ligand/From loaded ICM object - enter a_ligand.
- Click OK
- Docking/Run Docking Batch
- Once the docking has finished select Docking/Make Hitlist. Compare the docked ligand to the crystal structure ligand in 1PWM. The ligand should be accurately re-docked and overlay nicely with the crystal structure ligand. In the next part of this exercise we will try to determine if other inhibitors to this receptor could be identified using this one crystal structure or if more than one representation of the receptor is needed.
Step 4: Docking a Set of Known Inhibtors
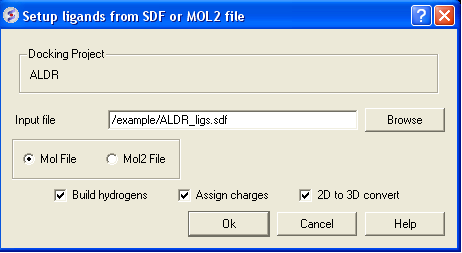
In the next part of this exercise we will try to determine if other inhibitors to this receptor could be correctly docked using this one crystal structure or if more than one representation of the receptor is needed. We have already setup the docking project and made all the required maps (Steps 1 to 3) so now let us dock a set of inhibitors to the receptor. We will dock a database called ALDR_ligs.sdf which should be present in your ICM distribution if you cannot find this example sdf file then please E mail support@molsoft.com and we will E mail it to you.
- Docking/Setup Batch Ligand/ From FIle: SDF/MOL2 - Select Mol and Check All boxes
- Docking/Run Docking Batch
- Once the docking has finished select Docking/Make Hitlist. Notice the poor poses and scores for some of the inhibitors - can these be improved by incorporating multiple receptor conformations?
Step 5: Generating Multiple Receptor Conformations
Here we will generate multiple receptor conformations of a flexible loop in the binding pocket of 1PWM.
- If you have viewed some previous docking results make sure the ligand is deleted from the object called ALDR_rec. You can do this by right click on the ligand name (most likely the third molecule in the object ALDR_rec).
- Expand the sequence display for the receptor in the ICM Workspace and select the loop region "CALLSC" (it is located towards the C-terminal). See screenshot below. You can select the region by clicking and dragging over the sequence in the ICM Workspace.

- MolMechanics/Sample Loop and check the boxes as shown below. This may take some time to run depending on the speed of your machine.
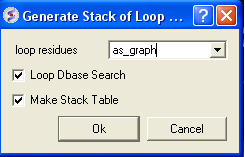
- Once the loop sampling has finished a table called confStack will be displayed. You can view the loop conformations by clicking on the rows.
Step 6: Docking to Multiple Receptor Conformations
- Delete all but the top 4 conformations in confStack. This can be done by clicking on the row header (5) and this will highlight the row in blue and then whilst holding the Shift key click on the header of the final row. Next right click on the row header and select and select Delete Rows.
- If you have viewed some previous docking results make sure the ligand is deleted from the object called ALDR_rec. You can do this by right click on the ligand name (most likely the third molecule in the object ALDR_rec).
- Docking/Flexible Receptor/Setup 4D grid and click OK.
- Docking/(Re)Make Receptor Maps
- Re-run docking batch /Docking/Run Docking Batch but change output file suffix to something different such as flex3conf.
- Once the docking has finished make the hitlist Docking/Make Hitlist.
- Browse the hitlist by clicking on the hitlist table and watch the receptor conformation switching along wih the ligand and note the additional column rec.conformation in the list. This column indicates which receptor in the stack the ligand is docked to.
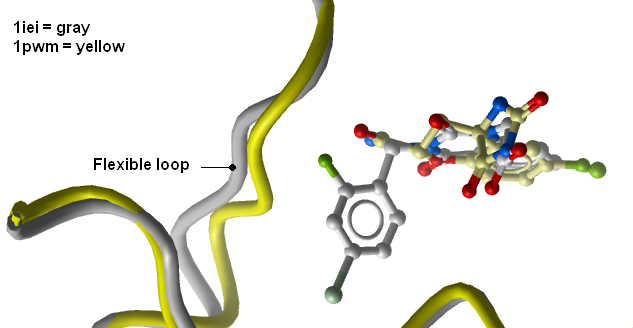
- Load PDB entry 1iei and compare the pose of the first ligand in the hit list and the corresponding receptor conformation to the experimental data.
- You will see that the flexibility in the loop region is accounted for by multiple receptor conformation docking and therefore inhibitors which scored poorly in Step 4 now score well and the corresponding receptor loop conformation for a ligand is identified.
| Prev Docking a Markush Generated Library | Home Up | Next Explicit Group |